BioinfinvRepro
Curso de introducción a la bioinformática e investigación reproducible
Análisis genético de poblaciones
Tabla de contenidos
- Tabla de contenidos
- Acerca de este tutorial
- Descripción de los datos
- Ubicación de los datos
- Generar un directorio de trabajo
- Paso 1
- Paso 2
- Paso 3: Filtrado de SNPs
- Paso 4: Borrar SNPs por filtro de HWE
- Paso 5: Eliminar parentescos desconocidos
- Paso 1: Merge de los datos ChileGenomico con 1000G
- Paso 2: Homologar la versión del genoma
- Paso 3: Fusionar los conjuntos de datos HapMap y 1000 Genomes
- Paso 4: Intercambiar alelos de SNPs con potenciales problemas de hebra
- Paso 5: Combinar 1000G con ChileGenomico.
- Paso 1: Realizar MDS en datos HapMap-ChileGenomico
- Paso 2: Generar un archivo con información de poblaciones
- Paso 3: Graficar resultados de MDS
- Paso 4: Realizar un análisis de ancestría
- Paso 5: Genere gráficos de sus resultados
Acerca de este tutorial
El objetivo del tutorial es introducir técnicas de el análisis de datos de genotipificación a nivel genómico para detectar estructura población. Se presenta un flujo de trabajo de análisis completo utilizando el comandos en UNIX y lenguaje R. La primera parte de este tutorial se base en el tutorial de GWA desarrollado independientemente por Andries Marees. Para más detalles ver el paper que acompaña a dicho tutorial GWA tutorial paper. Aunque se utilizan varios paquetes de R durante el tutorial, no se puede cubrir toda la funcionalidad disponible en cada uno de ellos. Consulte la documentación de cada software y paquete para obtener más detalles sobre su funcionalidad. El contenido de este tutorial es responsabilidad exclusiva del autor y no ha sido respaldado por los autores de los softwares ni paquetes de R aquí utilizados.
Descripción de los datos
Este tutorial utilizará datos públicos de genotipificación generados con microarreglos en los proyectos HapMap y ChileGenomico. Los datos en el tutorial de GWA y fueron obtenidos originalmente del sitio de NCBI.
Ubicación de los datos
Se utilizarán los datos del proyecto 1000G incluidos en el tutorial GWA_tutorial obtenido de este repositorio en GitHub. Puede encontrar el tutorial completo y los datos en el siguiente directorio:
/datos/compartido/GWA_tutorial/
Genere una variable de ambiente $G para almacenar la ruta a los datos de 1000G. Esto servirá para acortar los siguientes comandos.
$ export T=/datos/compartido/GWA_tutorial/1_QC_GWAS
$ export P=/datos/compartido/GWA_tutorial/2_Population_stratification/
nota 1: el símbolo ~ es un atajo para el directorio de origen del usuario (home). En este caso, /shared/bioinfo1.
nota 2: es importante que no existan espacios al rededor del símbolo igual. Si la tuviera espacios, hay que usar comillas dobles.
Los datos de ChileGenomico están ubicados en
/datos/compartido/ChileGenomico/
Genere una variable de ambiente para que sea más fácil acceder sus archivos.
$ export C=/datos/compartido/ChileGenomico
Los datos de 1000G están ubicados en
~/1000G/
Genere una variable de ambiente para que sea más fácil acceder sus archivos.
$ export G=~/1000G
Los scripts generados específicamente pare este tutorial se encuentran en
~/WK2019-PopGeno
Genere una variable de ambiente
$ export W=~/WK2019-PopGeno
Generar un directorio de trabajo
Genere un directorio para realizar su trabajo usando la inicial de su nombre y su apellido:
$ mkdir pguallardo
$ cd pguallardo
Genere un directorio dentro de este, para la realización de este tutorial:
$ mkdir -p Unidad2/Sesion2
$ cd Unidad2/Sesion2
Cargar el software
Para correr programas que no vienen con el sistema, primero debe cargar el módulo respectivo. En este tutorila usariemos Plink v1.9 y R v4.0.5
$ module load plink/1.90
$ module load R/4.0.5
Parte 1: Análisis de control de calidad
Paso 1
Investigar la cantidad de genotipos perdidos los datos de 1000G.
$ plink --bfile $C/chilean_all48_hg19 --missing
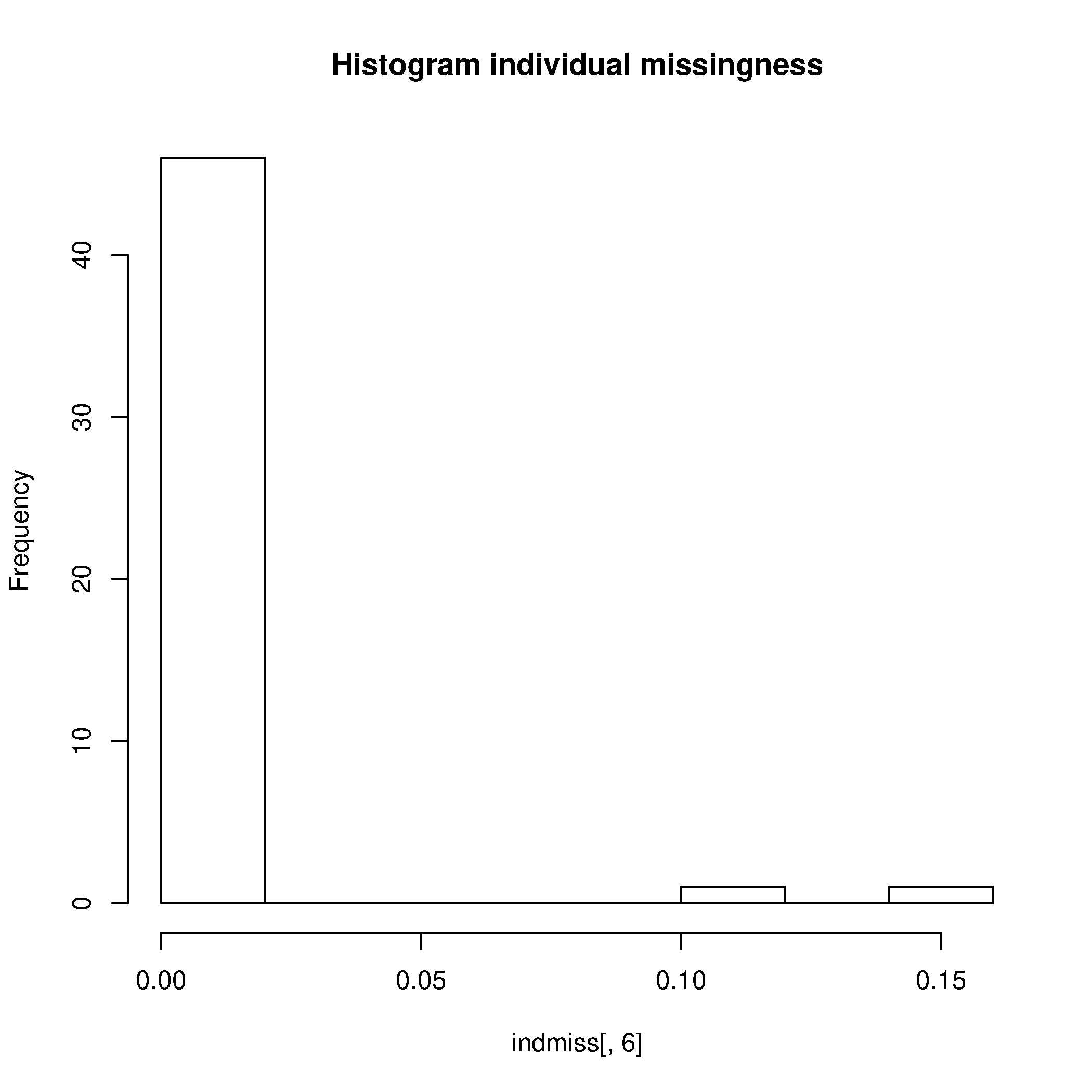
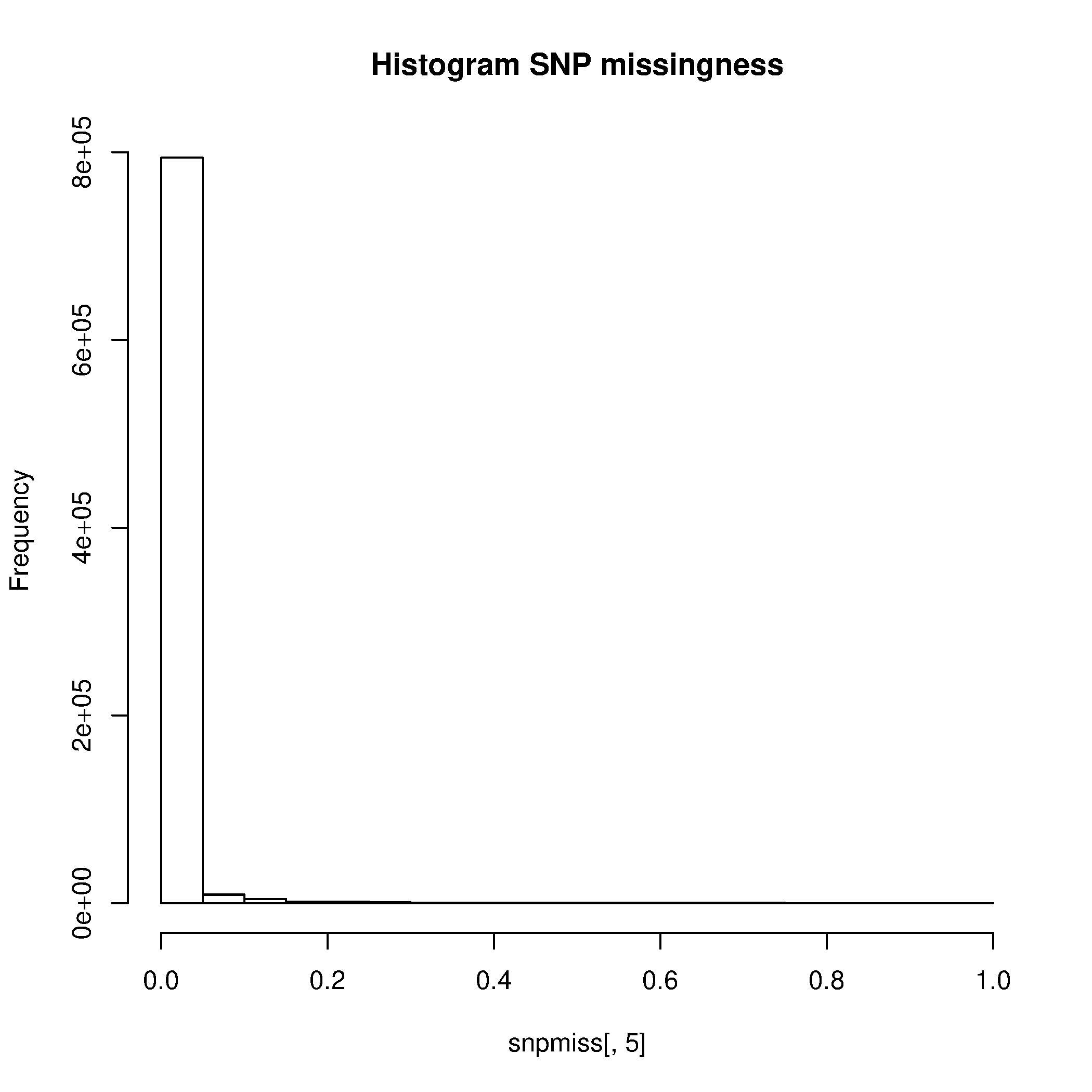
Este paso generó los siguientes archivos: plink.imiss y plink.lmiss, que muestras la proporción de datos perdidos para cada SNP y para cada y individuo, respectivamente.
Genere gráficos para visualizar los resultados
$ Rscript --no-save $T/hist_miss.R
Elimine los SNPs e individuos con alto nivel de datos perdidos (>0.02). Se puede encontrar la explicación para este paso y subsecuentes de control de calidad en el box 1 del paper citado y con los comentarios del tutorial asociado.
Elimine SNPs e individuos con pérdida de datos >0,2
plink --bfile $C/chilean_all48_hg19 --geno 0.2 --make-bed --out chilean_all48_hg19_2
plink --bfile chilean_all48_hg19_2 --mind 0.2 --make-bed --out chilean_all48_hg19_3
Elimine SNPs e individuos con pérdida de datos >0,02
plink --bfile chilean_all48_hg19_3 --geno 0.02 --make-bed --out chilean_all48_hg19_4
plink --bfile chilean_all48_hg19_4 --mind 0.02 --make-bed --out chilean_all48_hg19_5
Tarea:
Responda las siguientes preguntas:
- ¿Cómo se llaman los archivos que contienen las tasas de datos perdidos por SNP y por muestra?
- ¿Cuántas variantes se eliminaron por tener una tasa de datos perdidos mayor a 0.2?
- ¿Cuántos individuos tenían una tasa de datos perdidos mayor a 0.02?
- Basados en los histogramas y en sus cálculos, ¿qué valores umbrales de datos perdidos para muestras y SNPs sugeriría?
Paso 2
Revise discrepancias entre entre el sexo declarado en el la tabla de fenotipos (archivo ped/bed) y el inferido desde los genotipos.
Calcularemos es valor F, que se basa la estimación del coeficiente de consanguinidad (homocigocidad) en el cromosoma X. Los individuos de sexo femenino deberían tener un valor de F<0.2. Los de sexo masculino, >0,8. El programa PLINK etiquetará a los individuos que no cumplan con ninguna de estas dos condiciones con la palabra “PROBLEM”.
$ plink --bfile chilean_all48_hg19_5 --check-sex
Genere gráficos para visualizar el resultados del check de sexo.
$ Rscript --no-save $T/gender_check.R


Podemos ver que hay una mejor con una discrepancia de sexo, dado que tiene un valor de F de 0,99. Este tipo de problemas es usual cuando se analizan nuevos sets de datos, que aun no han sido curados. Generalmente están indicando un error en la identificación de muestras. Posiblemente se intercambió la muestra de un hombre con la de una mujer. Note que los cambios de muestras entre individuos del mismo sexo pasarán desapercibidos a esta prueba.
Visualize las primeras líneas del archivo generado.
$ head plink.sexcheck
FID IID PEDSEX SNPSEX STATUS F
CDSJ177 CDSJ177 1 1 OK 0.9147
CDSJ021 CDSJ021 1 1 OK 0.8743
ARI006 ARI006 1 1 OK 0.8936
ARI021 ARI021 1 1 OK 0.885
ARI022 ARI022 2 1 PROBLEM 0.9051
CDSJ174 CDSJ174 1 1 OK 0.8754
CDSJ175 CDSJ175 1 1 OK 0.8939
CDSJ046 CDSJ046 1 1 OK 0.8902
CDSJ176 CDSJ176 1 0 PROBLEM 0.7645
Genere una lista con los individuos con discrepancia de sexo. Este debe tener dos columnas, con el ID de familia y de individuo respectivamente.
$ grep "PROBLEM" plink.sexcheck | awk '{print$1,$2}'> sex_discrepancy.txt
Elimine los individuos con discrepancias
$ plink --bfile chilean_all48_hg19_5 --remove sex_discrepancy.txt --make-bed --out chilean_all48_hg19_6
Tarea:
- ¿Cuántos individuos fueron eliminados por discrepancia de sexo?
- ¿Qué riesgo(s) se corre(n) si no se eliminaran?
Paso 3: Filtrado de SNPs
Genere un archivo con SNPs autosomales solamente.
$ awk '{ if ($1 >= 1 && $1 <= 22) print $2 }' chilean_all48_hg19_6.bim > snp_1_22.txt
$ plink --bfile chilean_all48_hg19_6 --extract snp_1_22.txt --make-bed --out chilean_all48_hg19_7
Calcule las frecuencias alélicas para cada SNP.
$ plink --bfile chilean_all48_hg19_7 --freq --out MAF_check
Revise las primeras filas del archivo generado.
head MAF_check.frq
CHR SNP A1 A2 MAF NCHROBS
1 rs4951929 C T 0.08889 90
1 rs12562034 A G 0.1667 90
1 rs2980300 T C 0.2111 90
1 rs61768207 A G 0.05556 90
1 rs2518996 G A 0.4556 90
1 rs57181708 G A 0.1667 90
1 rs4422949 G A 0.2222 90
1 rs11516185 G A 0.4889 90
1 rs11507767 G A 0.1333 90
Genere a gráfico de la distribución de frecuencia del alelo menor (MAF por su sigla en inglés Mino Allele Frequency).
$ Rscript --no-save $T/MAF_check.R
Elimine los SNPs monomórficos (sin variabilidad). En el set hay, en este punto, 45 individuos, por lo tanto, la menor frecuencia alélica posible para un polimorfismo es 1/(2x45)=0,011. Utilizaremos este valor como mínima MAF. ** Nota: ** En el tutorial de GWAS además se borran los SNPs con frecuencia alélica menor a 0,05. Sin embargo eso no es necesario para estudios de estructura.
plink --bfile chilean_all48_hg19_7 --maf 0.011 --make-bed --out chilean_all48_hg19_8
...
Calculating allele frequencies... done.
98544 variants removed due to minor allele threshold(s)
(--maf/--max-maf/--mac/--max-mac).
459378 variants and 45 people pass filters and QC.
...
done.
Como ve, se eliminaron 98.544 SNPs por el filtro de frecuencia alélica.
Tarea
- ¿Cuál es el nombre del primer conjunto de datos que solo contiene SNPs en autosomas?
- ¿Cuántos SNPs se encontraban en cromosomas sexuales?
- ¿Como calcularía el número de cromosomas que porta cada uno de los alelos para cada SNP?
Paso 4: Borrar SNPs por filtro de HWE
Los SNPs cuyos genotipos tengan frecuencias que se desvíen demasiado desde los valores esperados en Equilibrio de Hardy-Weinberg (HWE) pueden tener errores de genotipificación y deben ser eliminados.
Revise la distribución de valores de p para el test HWE.
plink --bfile chilean_all48_hg19_8 --hardy
vamos a seleccionar SNPs con valores p < 0.000001, lo cual es requerido por unos de los dos siguientes scripts de R. Esto nos permitirá visualizar los SNPs con grandes de desvíos desde HWE.
awk '{ if ($9 <0.000001) print $0 }' plink.hwe > plinkzoomhwe.hwe
Rscript --no-save $T/hwe.R
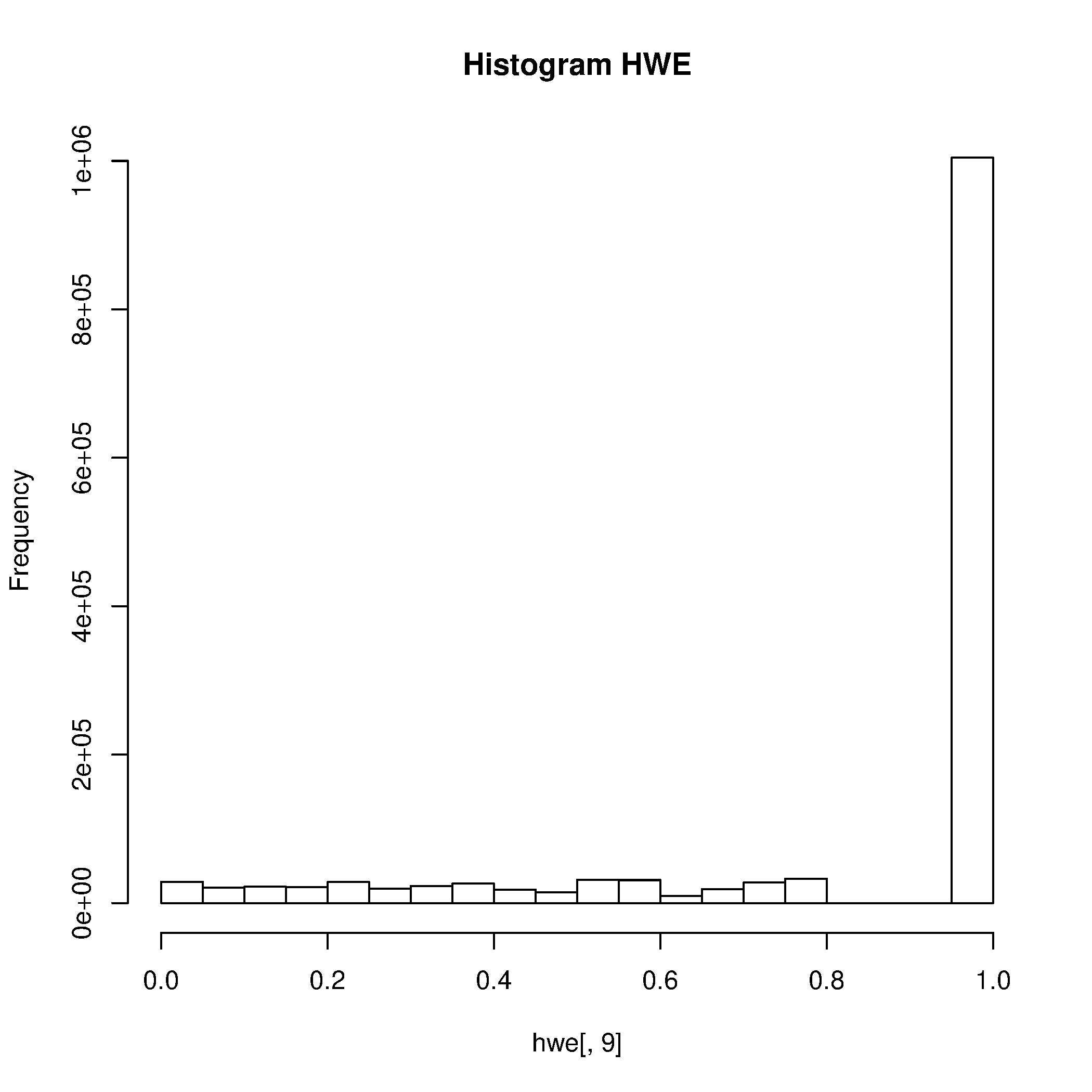
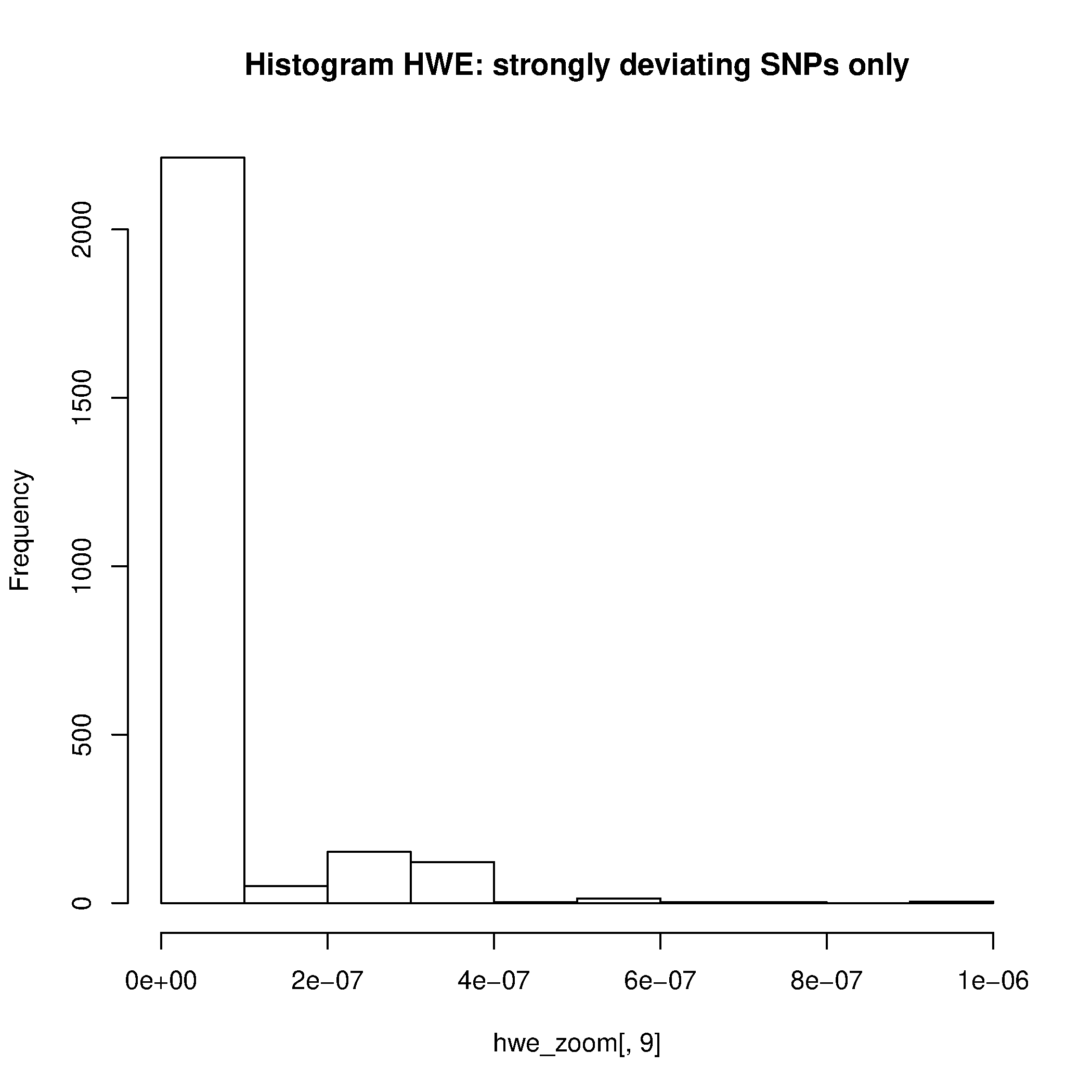
Filtre los SNPs que no cumplen con el filtro MAX>0,000001.
plink --bfile chilean_all48_hg19_8 --hwe 1e-6 --hwe-all --make-bed --out chilean_all48_hg19_9
* Nota:* en el tutorial de GWA se eliminan individuos con heterocigocidad mayor o menor a 3 desviaciones estándar desde el promedio, dado que sugieren mezcla de muestras o consanguinidad. No está claro que este criterio sea válido poblaciones mestizas. Aquí saltaremos ese filtro, principalmente para ahorrar tiempo.
Tarea
- ¿Cuál es el nombre del archivo con los resultados de la prueba de HWE?
- ¿Basándose en la distribución de los valores de p, le parece el umbral usado razonable o propondría otro valor?
Paso 5: Eliminar parentescos desconocidos
Es muy importante revisar si existen parentesco desconocidos en el set de datos, porque pueden sesgar las estimaciones de ancestría (y de asociación con fenotipos).
Las pruebas de parentesco asumen que los SNP no están correlacionados, vale decir que no tienen un fuerte ligamiento. Para generar una lista de SNPs sin correlaciones muy altas, exploremos regiones de inversión (inversion.txt [regiones con alto LD]) y eliminaremos SNPs con el argumento command --indep-pairwise.
Los parámetros 50 5 0.2 indican: tamaño de ventana en número de SNPs, el paso de desplazamiento de la ventana en número de SNPs, y el máximo coeficiente de correlación múltiple (r2) de un SNP respecto de todos los otros SNPs en la ventana.
plink --bfile chilean_all48_hg19_9 --exclude $T/inversion.txt --range --indep-pairwise 50 5 0.2 --out indepSNP
Asumiendo una muestra poblacional aleatoria, vamos a excluir todos los individuos que tengan un valor pihat ≥ 0,2.
plink --bfile chilean_all48_hg19_9 --extract indepSNP.prune.in --genome --min 0.2 --out pihat_min0.2
Visualice las primeras líneas del resultado.
$ head pihat_min0.2.genome | cut -c 1-77
FID1 IID1 FID2 IID2 RT EZ Z0 Z1 Z2 PI_HAT
CDSJ177 CDSJ177 ARI001 ARI001 UN NA 0.4733 0.5267 0.0000 0.2634
CDSJ021 CDSJ021 ARI001 ARI001 UN NA 0.5359 0.4641 0.0000 0.2320
ARI006 ARI006 ARI001 ARI001 UN NA 0.4314 0.5686 0.0000 0.2843
ARI021 ARI021 ARI018 ARI018 UN NA 0.6915 0.1554 0.1530 0.2307
ARI021 ARI021 ARI015 ARI015 UN NA 0.6506 0.2759 0.0736 0.2115
ARI021 ARI021 ARI001 ARI001 UN NA 0.4205 0.5795 0.0000 0.2897
ARI021 ARI021 ARI019 ARI019 UN NA 0.6748 0.2174 0.1078 0.2165
CDSJ174 CDSJ174 ARI001 ARI001 UN NA 0.5196 0.4804 0.0000 0.2402
CDSJ175 CDSJ175 ARI001 ARI001 UN NA 0.4559 0.5441 0.0000 0.2720
$ head pihat_min0.2.genome | cut -c 78-
PHE DST PPC RATIO
-1 0.788650 1.0000 3.6279
-1 0.773401 1.0000 3.5860
-1 0.787614 1.0000 4.2789
-1 0.848091 1.0000 2.6656
-1 0.841702 1.0000 3.1995
-1 0.792681 1.0000 4.1797
-1 0.843969 1.0000 2.8519
-1 0.772496 1.0000 3.4360
-1 0.788864 1.0000 3.8086
Genere gráficos para evaluar estos parentesco con el siguientes script en R.
Rscript --no-save $T/Relatedness.R
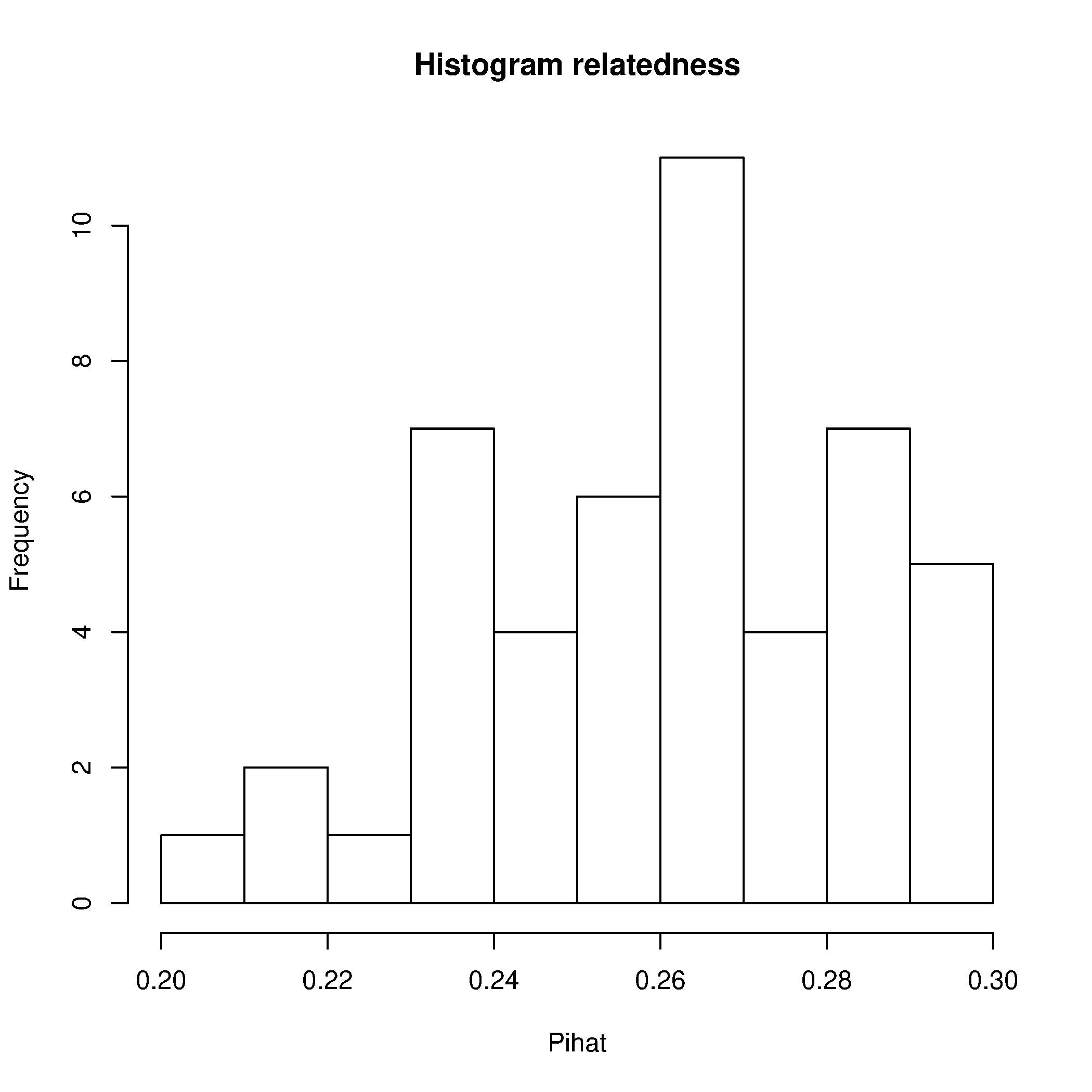
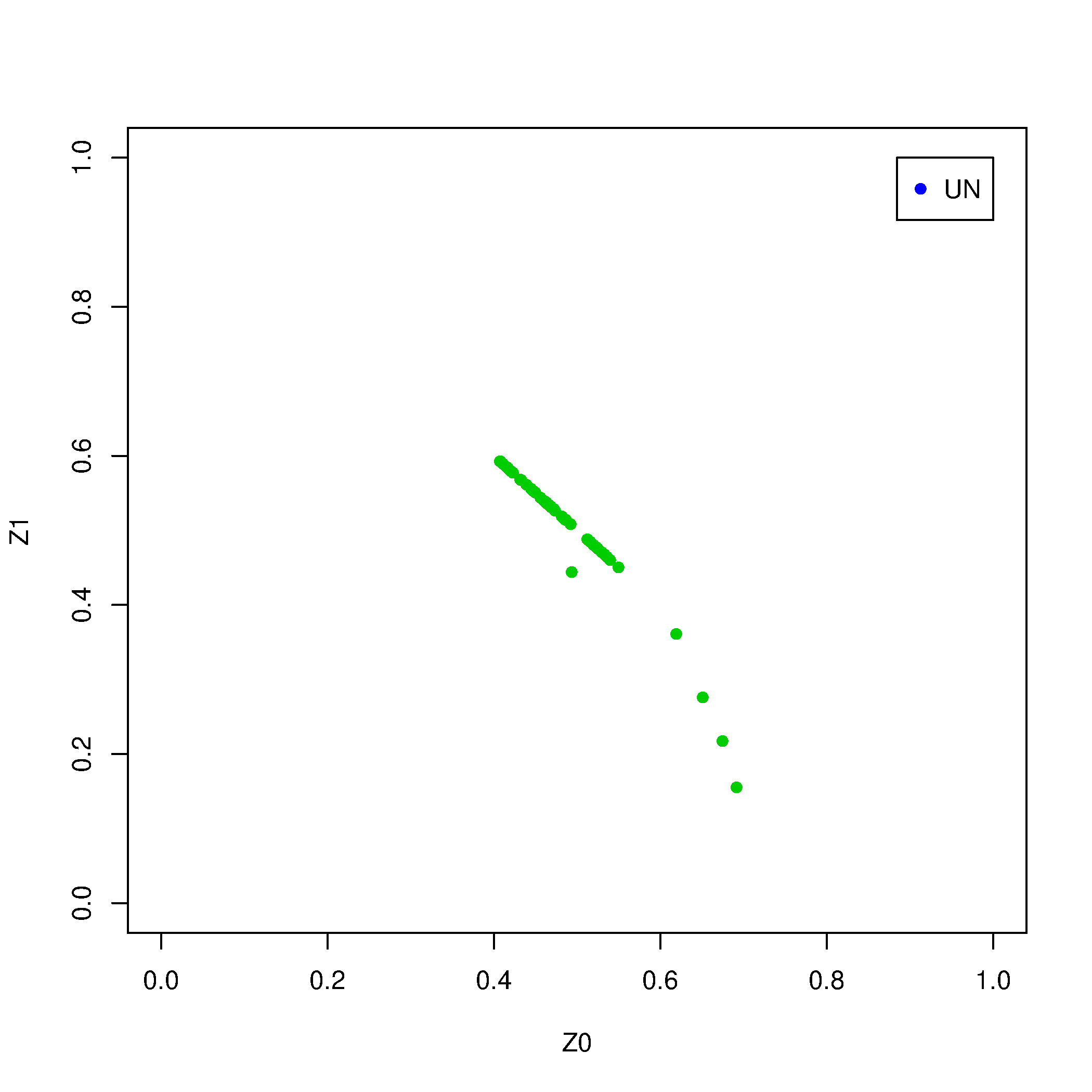 Explicación: PO = parent-offspring, UN = unrelated individuals.
Explicación: PO = parent-offspring, UN = unrelated individuals.
Usemos R para investigar cuáles son los individuos que están generando más problemas.
$ R
> rel <- read.table("pihat_min0.2.genome", header=T)
> table(rel$IID1)
ARI001 ARI002 ARI003 ARI004 ARI006 ARI008 ARI013 ARI015 ARI017 ARI018 ARI021
11 1 1 1 1 1 1 1 1 2 4
ARI023 CDSJ020 CDSJ021 CDSJ046 CDSJ099 CDSJ122 CDSJ157 CDSJ174 CDSJ175 CDSJ177 CDSJ262
1 1 1 1 1 1 1 1 1 1 1
CDSJ283 CDSJ284 CDSJ297 CDSJ299 CDSJ347 CDSJ361 CDSJ372 CDSJ400 CDSJ403 CDSJ469 CDSJ471
1 1 1 1 1 1 1 1 1 1 1
CDSJ472
1
> table(rel$IID2)
ARI001 ARI008 ARI014 ARI015 ARI018 ARI019 CDSJ032 CDSJ048 CDSJ106 CDSJ108 CDSJ167
32 1 2 1 1 3 1 1 1 1 1
CDSJ321 CDSJ344 CDSJ417
1 1 1
Claramente el individuo ARI001 genera la mayoría de los aparentes parentescos. Revisemos qué parentescos quedan si eliminamos este individuo.
> subset(rel, !IID1 %in% "ARI001" & !IID2 %in% "ARI001")
FID1 IID1 FID2 IID2 RT EZ Z0 Z1 Z2 PI_HAT PHE DST PPC
4 ARI021 ARI021 ARI018 ARI018 UN NA 0.6915 0.1554 0.1530 0.2307 -1 0.848091 1
5 ARI021 ARI021 ARI015 ARI015 UN NA 0.6506 0.2759 0.0736 0.2115 -1 0.841702 1
7 ARI021 ARI021 ARI019 ARI019 UN NA 0.6748 0.2174 0.1078 0.2165 -1 0.843969 1
16 ARI018 ARI018 ARI014 ARI014 UN NA 0.4936 0.4438 0.0626 0.2845 -1 0.852064 1
48 ARI008 ARI008 ARI019 ARI019 UN NA 0.6189 0.3611 0.0200 0.2005 -1 0.837686 1
RATIO
4 2.6656
5 3.1995
7 2.8519
16 4.0182
48 3.3516
El individuo ARI021 y el ARI018 también son candidatos a ser eliminados. Sin bien ARI008 y ARI019 tienen un coeficiente de parentesco de 0.2005, está muy cerca del límite que queremos aceptar así que los dejaremos. Además, hay que considerar que los coeficientes de parentesco pueden estar sobre estimados en individuos mestizos. El software REAP puede dar cuenta de esto y generar mejores estimaciones.
Cree un archivo con el ID de familia y de individuo para ser eliminado usando el editor de texto vi.
$ awk '$2=="ARI001" || $2=="ARI021" || $2=="ARI018"' chilean_all48_hg19_9.fam > to_romeve_by_relatedness.txt
Borre los individuos (en este caso solo uno) con la menor tasa de genotipificación por cada par con pihat > 0,2 .
$ plink -bfile chilean_all48_hg19_9 -remove to_romeve_by_relatedness.txt -make-bed --out chilean_all48_hg19_10
Tarea
- ¿Cuántos SNPs en aparente equilibrio de ligamiento se encontraron?
- ¿Cuántos SNPs se eliminaron por estar en regiones de inversiones conocidas?
- ¿Cuántos individuos quedaron luego del filtro de parentesco?
- ¿Cuál fue el mayor coeficiente de parentesco efectivamente aceptado?
Parte 2: Unir datos locales con 1000G
Para esta parte del tutorial, necesitará los archivo filtrados generados anteriormente: chilean_all48_hg19_10.fam, chilean_all48_hg19_10.bim, chilean_all48_hg19_10.bed, y el archivo de SNPs con bajo LD: indepSNP.prune.in.
Adicionalmente usaremos datos obtenidos del proyecto 1000G. El preprocesamiento de los datos y control de calidad ya fueron realizados, siguiendo los pasos previamente descritos. Los datos y el script de comandos usados se encuentran en ``.
Paso 1: Merge de los datos ChileGenomico con 1000G
Elimine variantes duplicadas del set ChileGenomico
$ plink --bfile chilean_all48_hg19_10 --list-duplicate-vars suppress-first
$ plink --bfile chilean_all48_hg19_10 --exclude plink.dupvar --make-bed --out chilean_all48_hg19_11Extraiga las variantes presentes en los datos de ChileGenomico.
$ cut -f 2 chilean_all48_hg19_11.bim | sort -u > chilean_all48_hg19_11.snps
* Nota:* usamos un pipe para order la lista de snps dado que es un requisito para el siguiente comando.
Extraiga las variantes presentes en los datos de 1000G.
$ cut -f 2 $G/1kG_MDS5.bim | sort -u > 1kG_MDS5.snps
Nota: Usamos $G para buscar el set 1kG_MFS5.bim en su carpeta en vez de copiarla al directorio de trabajo.
Encuentre la lista de SNPs en común entre ambos sets de datos.
$ comm -12 chilean_all48_hg19_11.snps 1kG_MDS5.snps > common_snps.txt
Extraiga los SNPs en común del set 1000G
$ plink --bfile $G/1kG_MDS5 --extract common_snps.txt --recode --make-bed --out 1kG_MDS6
Extraiga los SNPs en común del set ChileGenomico
$ plink --bfile chilean_all48_hg19_11 --extract common_snps.txt --make-bed --out chilean_all48_hg19_12
Los conjuntos de datos ahora contienen exactamente las mismas variantes.
Paso 2: Homologar la versión del genoma
Los conjuntos de datos deben tener la misma versión de ensamble del genoma para usar las mismas coordenadas de SNPs. Para asegurarnos, cambiaremos las coordenadas en los datos de 1000 genomas usando las coordenadas de ChileGenomico.
$ awk '{print $2, $4}' chilean_all48_hg19_12.bim> buildhapmap.txt
buildhapmap.txt contiene un ID y una posición física por SNP en cada línea.
$ plink --bfile 1kG_MDS6 --update-map buildhapmap.txt --make-bed --out 1kG_MDS7
chilean_all48_hg19_12 y 1kG_MDS7 ahora tienen las mismas coordenadas.
Paso 3: Fusionar los conjuntos de datos HapMap y 1000 Genomes
Antes de fusionar los datos de ChileGenomico con los datos de HapMap, queremos asegurarnos de que los archivos puedan fusionarse, para ello realizamos 3 pasos:
1) Asegurarse de que el genoma de referencia sea igual en ambos conjuntos de datos 1kG y ChileGenomico (que usen el mismo alelo A1 de referencia en el archivo bim). 2) Resolver problemas de hebra. 3) Eliminar los SNP que, después de los dos pasos anteriores, todavía difieren entre los conjuntos de datos.
Los siguientes pasos pueden ser bastante técnicos en términos de comandos, pero solo comparamos los dos conjuntos de datos y nos aseguramos de que correspondan.
1) establecer el genoma de referencia en ChileGenomico usando 1000G:
$ awk '{print $2, $5}' 1kG_MDS7.bim> 1kG_ref-list.txt
$ plink --bfile chilean_all48_hg19_12 --reference-allele 1kG_ref-list.txt --make-bed --out chilean_all48_hg19_13
Los conjuntos de datos 1kG y chilean_all48_hg19_13 tienen el mismo genoma (alelo) de referencia para todos los SNP.
Este comando podría generar algunas advertencias para la asignación imposible del alelo A1. Es bueno revisar:
$ grep "Impossible" chilean_all48_hg19_13.log | wc -l
0
En este caso no se encontraron inconsistencias. Si la hubiere, podría indicar errores de genotipificación o el uso de sondas distintas (si ambos sets provenieran de microarreglos) que quizás estén interrogando distintas hebras.
2) Resolver problemas de hebra. Compruebe si hay posibles problemas de filamento.
$ awk '{print $2, $5, $6}' 1kG_MDS7.bim> 1kG_MDS7_tmp
$ awk '{print $2, $5, $6}' chilean_all48_hg19_13.bim > chilean_all48_hg19_13_tmp
$ sort 1kG_MDS7_tmp chilean_all48_hg19_13_tmp | uniq -u > all_differences.txt
$ wc -l all_differences.txt
0 all_differences.txt
En este caso hay 0 diferencias entre los archivos. Si las hubiera, podrían deberse a problemas cambio de hebra entre conjuntos de datos.
Paso 4: Intercambiar alelos de SNPs con potenciales problemas de hebra
Este paso no tendrá efecto alguno sobre los datos porque no hay SNPs con problemas de hebra. Sin embargo, los incluimos en nuestro flujo de trabajo ya que pueden tener efecto en un conjunto de datos distinto.
Imprima el identificador SNP y elimine los duplicados.
$ awk '{print $ 1}' all_differences.txt | sort -u > flip_list.txt
Se genera un archivo de SNPs no coincidentes entre los dos archivos.
Intercambiar los alelos de los SNPs no coincidentes.
$ plink --bfile chilean_all48_hg19_13 --flip flip_list.txt --reference-allele 1kG_ref-list.txt --make-bed --out chilean_all48_hg19_14
Compruebe si hay SNPs que siguen siendo problemáticos después de haberlos volteado.
$ awk '{print $2, $5, $6}' chilean_all48_hg19_14.bim > chilean_all48_hg19_14_tmp
$ sort 1kG_MDS7_tmp chilean_all48_hg19_14_tmp | uniq -u> uncorresponding_SNPs.txt
$ wc -l uncorresponding_SNPs.txt
0 uncorresponding_SNPs.txt
Este archivo demuestra que hay 0 diferencias entre los archivos, o sea, no hay SNPs con alelos discordantes entre conjuntos de datos.
3) Eliminar SNPs problemáticos de 1kG y ChileGenomico. En este caso, este paso es innecesario ya que no quedan SNPs con problemas, pero se muestran por completitud.
$ awk '{print $ 1}' uncorresponding_SNPs.txt | sort -u > SNPs_for_exlusion.txt
El comando anterior genera una lista de los 0 SNP que causaron las 0 diferencias entre los conjuntos de datos 1kG y ChileGenomico después de cambiar y configurar el genoma de referencia.
Elimine los SNPs problemáticos de ambos conjuntos de datos.
$ plink --bfile 1kG_MDS7 --exclude SNPs_for_exlusion.txt --make-bed --out 1kG_MDS8
$ plink --bfile chilean_all48_hg19_14 --exclude SNPs_for_exlusion.txt --make-bed --out chilean_all48_hg19_15
Paso 5: Combinar 1000G con ChileGenomico.
$ plink --bfile 1kG_MDS8 --bmerge chilean_all48_hg19_15.bed chilean_all48_hg19_15.bim chilean_all48_hg19_15.fam --allow-no-sex --make-bed --out MDS_merge
Parte 3: Análisis de estructura poblacional
Paso 1: Realizar MDS en datos HapMap-ChileGenomico
Se asume que los SNPs no están ligados. Por lo tanto usaremos un conjunto de SNPs con bajo LD
$ plink --bfile MDS_merge --extract indepSNP.prune.in --genome --out MDS_merge2
$ plink --bfile MDS_merge --read-genome MDS_merge2.genome --cluster --mds-plot 10 --out MDS_merge2
* Nota*: esto no reduce el tamaño del conjunto de datos MDS_merge2, solo crea el archivo MDS_merge2.genome para el set reducido de SNPs con bajo LD.
Paso 2: Generar un archivo con información de poblaciones
El archivo $G/20100804.ALL.panel (obtenido desde aquí) contiene códigos de población de los individuos de 1000 genomas .
Convierta los códigos de población en códigos de superpoblación (es decir, AFR, AMR, ASN y EUR).
awk '{print$1,$1,$2}' $G/20100804.ALL.panel > ethnicity_1kG.txt
sed 's/JPT/ASN/g' ethnicity_1kG.txt>ethnicity_1kG2.txt
sed 's/ASW/AFR/g' ethnicity_1kG2.txt>ethnicity_1kG3.txt
sed 's/CEU/EUR/g' ethnicity_1kG3.txt>ethnicity_1kG4.txt
sed 's/CHB/ASN/g' ethnicity_1kG4.txt>ethnicity_1kG5.txt
sed 's/CHD/ASN/g' ethnicity_1kG5.txt>ethnicity_1kG6.txt
sed 's/YRI/AFR/g' ethnicity_1kG6.txt>ethnicity_1kG7.txt
sed 's/LWK/AFR/g' ethnicity_1kG7.txt>ethnicity_1kG8.txt
sed 's/TSI/EUR/g' ethnicity_1kG8.txt>ethnicity_1kG9.txt
sed 's/MXL/AMR/g' ethnicity_1kG9.txt>ethnicity_1kG10.txt
sed 's/GBR/EUR/g' ethnicity_1kG10.txt>ethnicity_1kG11.txt
sed 's/FIN/EUR/g' ethnicity_1kG11.txt>ethnicity_1kG12.txt
sed 's/CHS/ASN/g' ethnicity_1kG12.txt>ethnicity_1kG13.txt
sed 's/PUR/AMR/g' ethnicity_1kG13.txt>ethnicity_1kG14.txt
Crea un archivo de etnicidad de los datos de ChileGenomico.
$ awk '{if($1~/CDSJ/) pop="MAP"}{if($1~/ARI/) pop="AYM"} {print $1, $2, pop}' chilean_all48_hg19_14.fam > ethnicityfile_CLG.txt
Concatenar los archivos de carrera.
$ cat ethnicity_1kG14.txt ethnicityfile_CLG.txt | sed -e '1i \ FID IID ethnicity'> ethnicityfile.txt
Paso 3: Graficar resultados de MDS
$ Rscript $W/MDS_merged.R
IID FID.x SOL C1 C2 C3 C4 C5
1 ARI001 ARI001 0 -0.0406769 -0.01424110 -0.0924081 0.001803800 -0.001256100
2 ARI002 ARI002 0 -0.0469810 -0.00404251 -0.0788820 0.001140350 -0.001596390
3 ARI003 ARI003 0 -0.0514636 -0.01349430 -0.0900590 0.000246449 0.000925401
4 ARI004 ARI004 0 -0.0615831 -0.02951150 -0.1095080 -0.003338710 -0.002250870
5 ARI006 ARI006 0 -0.0514338 -0.02318140 -0.1022530 -0.001913330 -0.002395160
6 ARI008 ARI008 0 -0.0521863 -0.01152950 -0.0921278 0.001553320 -0.000445462
C6 C7 C8 C9 C10 FID.y
1 0.004404190 -0.007447020 0.014184100 -0.010402400 0.00411825 ARI001
2 0.002151910 0.001896250 0.000439761 -0.004716690 0.00383263 ARI002
3 0.000133159 -0.000325179 0.004675980 -0.004088450 -0.00150137 ARI003
4 -0.000113890 0.000147362 -0.001522850 0.000552478 -0.00157933 ARI004
5 0.001440190 -0.003645160 0.000958471 -0.003207940 -0.00300836 ARI006
6 0.001110960 0.001800130 0.001531390 -0.004898150 -0.00331712 ARI008
ethnicity
1 AYM
2 AYM
3 AYM
4 AYM
5 AYM
6 AYM
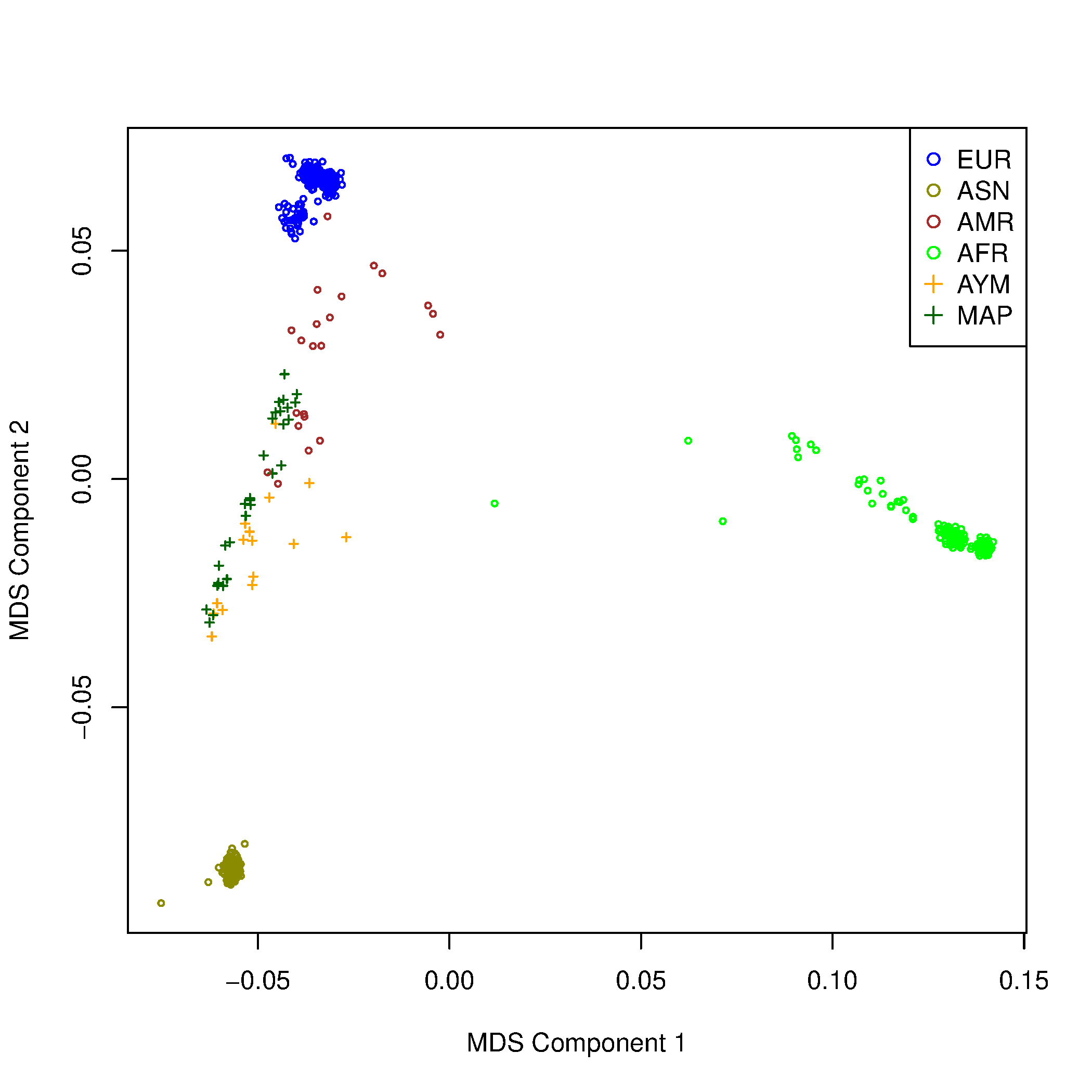
Tarea:
- En R, genere gráficos similares para las combinaciones Component 2 vs 3 y 3 vs 4. ¿Qué puede concluir de estos gráficos?
Paso 4: Realizar un análisis de ancestría
Utilizaremos la herramienta ADMIXTURE para inferir ancestría de las muestras chilenas asumiendo un modelo simple de mezcla.
Admixture asumen que los SNPs a usar no están en LD. Tendremos que usar nuevamente nuestro archivo con SNPs en bajo LD, pero esta vez sí crearemos un set plink de datos reducidos
$ plink --bfile MDS_merge --extract indepSNP.prune.in --make-bed --out MDS_merge_r2_lt_0.2
- ¿Cuántos SNPs quedaron luego del filtro?
- ADMIXTURE asume que los individuos no están emparentados. Sin embargo, no realizamos ningún filtro. ¿Por qué?
Ahora sí podemos correr ADMIXTURE.
$ module load admixture
$ admixture -j4 --cv MDS_merge_r2_lt_0.2.bed 3 > MDS_merge_r2_lt_0.2.K3.log
Los argumentos usados fueron:
-j4 usar cuatro hebras de cómputo
--cv realizar validación cruzada
MDS_merge_r2_lt_0.2.bed archivo en formad bed (plink)
3 número de poblaciones ancestrales (parámtro K)
> redirección de la salida a un archivo
MDS_merge_r2_lt_0.2.K3.log archivo log con la salida del programa
Repitamos el procesos para valores K de 4 a 6 usando un loop.
for k in $(seq 4 6)
do
admixture -j44 --cv MDS_merge_r2_lt_0.2.bed $k > MDS_merge_r2_lt_0.2.K$k.log
done
Tarea:
- ¿Cuántos SNPs quedaron luego del filtro?
- ADMIXTURE asume que los individuos no están emparentados. Sin embargo, no realizamos ningún filtro. ¿Por qué?
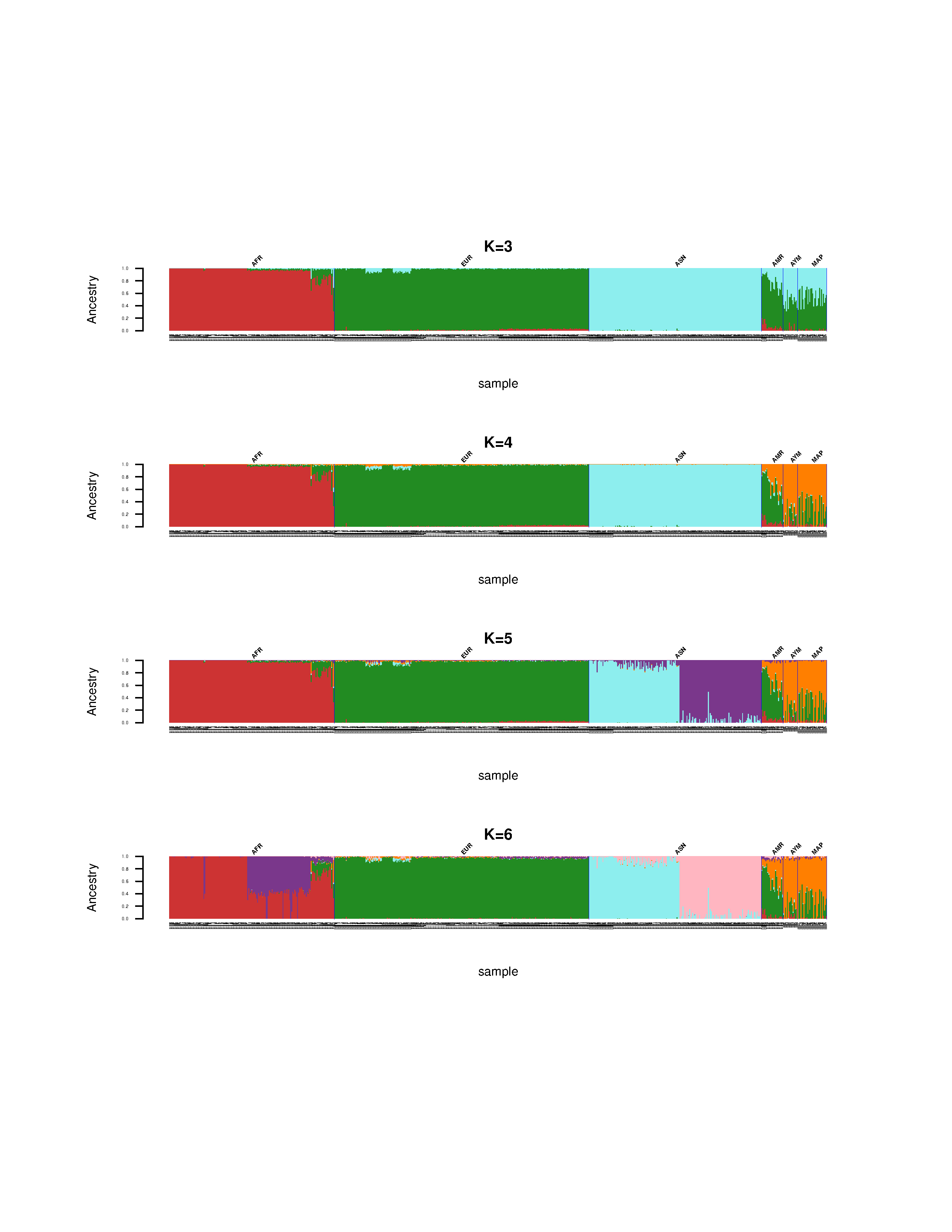
Paso 5: Genere gráficos de sus resultados
Primero tenemos que ordenar el archivo con la información de las muestras por públición. Esto es un requisito para el siguiente paso. Aquí elegiremos el orden en el que queremos que se muestren las poblaciones, mediante unos pocos comandos en R.
$ R --no-save
popinfo <- read.table("ethnicityfile.txt", as.is=T, header=T)
popinfo_ls <- split(popinfo, popinfo[,3])
names(popinfo_ls)
[1] "AFR" "AMR" "ASN" "AYM" "EUR" "MAP"
popinfo_sorted <- do.call(rbind, popinfo_ls[c("AFR", "EUR", "ASN", "AMR", "AYM", "MAP")])
write.table(popinfo_sorted, "popinfo_sorted.txt", sep="\t", row.names=F)
q()
Utilizaremos un script en R para traficar los resultados para todos los valores de K.
Rscript $W/admixture_plot.R popinfo_sorted.txt MDS_merge_r2_lt_0.2.fam