BioinfinvRepro
Curso de introducción a la bioinformática e investigación reproducible
Tutorial para el llamado de variantes
Karen Oróstica, Cristian Yáñez Lara y Ricardo Verdugo Creado abril 2020, actualizado noviembre 2025
El siguiente protocolo está diseñado para ser implementado en el servidor genoma.med.uchile.cl dentro de la carpeta de cada usuario de bioinfo1. Además, se asume que ya se ejecutó el tutorial Tutorial para el filtro y alineamiento de lecturas.
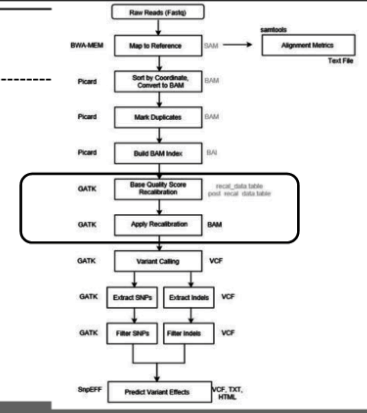
Procesamiento del alineamiento usando GATK
Usamos el programa Genome Analysis Toolkit (GATK) para el procesamiento en las siguientes etapas. De forma general los parámetros de GATK indican lo siguiente:
| Parámetro | Descripción |
|---|---|
| -T | Herramienta utilizada |
| -R | Archivo de referencia (el mismo usado en el alineamiento) |
| -I | Archivo de entrada el cual varía según la etapa del análisis |
| -L | Archivo .bed con las regiones target del experimento |
| -o | Archivo de salida generado el cual varía según la etapa del análisis |
Recalibración de calidades de bases (Q) en lecturas
La etapa de calibración de puntuación de calidad de base (BQSR) aplica un modelo de error empíricamente preciso a las bases.
Comando:
java -jar GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R reference.fasta \
-I muestra_sorted_RG.bam \
-knownSites dbSNP_hg19.vcf \
-o muestra_recal_data.table
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T BaseRecalibrator -R /datos/reference/genomes/hg19_reference/hg19.fasta -I S10_sorted_RG.bam -knownSites /datos/reference/genomes/hg19_reference/dbSNP_hg19.vcf -o S10_recall_data.table
Esto crea un archivo “muestra_recall_data.table” que contienen los datos de covarianza que se utilizarán en el paso posterior.
Nota: Es importante proporcionar a GATK un conjunto de sitios conocidos con el parámetro “-knownSites”, de lo contrario se negará a ejecutar el programa, en este caso se le proporciona las variantes de la base de datos dbSNP que posee GATK.
Aplicar la recalibración a los datos de secuencia
Comando:
java -jar GenomeAnalysisTK.jar \
-T PrintReads \
-R reference.fasta \
-I muestra_sorted_RG.bam \
--BQSR muestra_recal_data.table \
-o muestra_recal_reads.bam
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T PrintReads -R /datos/reference/genomes/hg19_reference/hg19.fasta -I S10_sorted_RG.bam --BQSR S10_recall_data.table -o S10_recall_reads.bam
Llamado de variantes
Identificar variantes
Usando HaplotypeCaller, un llamador de variantes de GATK se pueden identificar dos tipos de variantes a partir del alineamiento procesado: polimorfismos de un solo nucleótido (SNP) e inserción-deleción (InDels). Se obtienen las variantes almacenadas en un archivo “Variant Call Format” (VCF).
Comando:
java -jar GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R reference \
-I muestra_recal_reads.bam \
--dbsnp dbSNP_hg19.vcf \
-stand_call_conf 30 \
-L "chr19" \
-o muestra_raw_variants.vcf
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T HaplotypeCaller -R /datos/reference/genomes/hg19_reference/hg19.fasta -I S10_recall_reads.bam --dbsnp /datos/reference/genomes/hg19_reference/dbSNP_hg19.vcf -stand_call_conf 30 -L "chr19" -o S10_raw_variants.vcf
**Nota **: Para acelerar el tutorial solo se calculan las variantes en el cromosoma 19. Para analizar todos los cromosomas debe eliminar el parámetro -L “chr19” del comando.
Aplicar filtros a las variantes
El identificador de variantes GATK (HaplotypeCaller) es por diseño muy indulgente en las variantes de llamada para lograr un alto grado de sensibilidad. Esto es bueno porque minimiza la posibilidad de perder variantes reales, pero significa que debemos refinar el conjunto de llamadas para reducir la cantidad de falsos positivos, que puede ser bastante grande, lo que se hace en la etapa de filtrado de variantes.
Se ejecuta el proceso de filtrado por separado en SNPs e Indels, por lo que se dividen las variantes en archivos separados. Para SNPs e Indels se pueden aplicar los mismos filtros pero con distintos valores o parámetros. Para efectos prácticos solo filtraremos por profundidad de cobertura de las variantes.
Extraer SNPs
Extraemos los SNP del conjunto de variantes llamadas
Comando:
java -jar GenomeAnalysisTK.jar \
-T SelectVariants \
-R reference.fasta \
-V muestra_raw_variants.vcf \
-selectType SNP \
-o muestra_RAW_SNP.vcf
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T SelectVariants -R /datos/reference/genomes/hg19_reference/hg19.fasta -V S10_raw_variants.vcf -selectType SNP -o S10_RAW_SNP.vcf
Filtrar SNPs
Aplicamos el filtro al conjunto de SNPs. Usamos la expresión ‘DP> 10’ para incluir sólo los sitios con suficiente profundidad (depth > 10).
Comando:
java -jar GenomeAnalysisTK.jar \
-T VariantFiltration \
-R reference.fasta \
-V muestra_RAW_SNP.vcf \
--filterExpression "DP <10" \
--filterName "FILTER" \
-o muestra_FILTERED_SNP.vcf
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T VariantFiltration -R /datos/reference/genomes/hg19_reference/hg19.fasta -V S10_RAW_SNP.vcf --filterExpression "DP <10" --filterName "FILTER" -o S10_FILTERED_SNP.vcf
Extraer InDels
Extraemos los Indels del conjunto de variantes llamadas
Comando:
java -jar GenomeAnalysisTK.jar \
-T SelectVariants \
-R reference.fasta \
-V muestra_raw_variants.vcf \
-selectType INDEL \
-o muestra_RAW_INDEL.vcf
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T SelectVariants -R /datos/reference/genomes/hg19_reference/hg19.fasta -V S10_raw_variants.vcf -selectType INDEL -o S10_RAW_INDEL.vcf
Filtrar InDels
Aplicamos el filtro al conjunto de Indels
Comando:
java -jar GenomeAnalysisTK.jar \
-T VariantFiltration \
-R reference.fasta \
-V muestra_RAW_INDEL.vcf \
--filterExpression "DP <10" \
--filterName "FILTER" \
-o muestra_FILTERED_INDEL.vcf
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T VariantFiltration -R /datos/reference/genomes/hg19_reference/hg19.fasta -V S10_RAW_INDEL.vcf --filterExpression "DP <10" --filterName "FILTER" -o S10_FILTERED_INDEL.vcf
Nota: Las variantes que pasan los filtros se etiquetan como PASS y las que no pasan los filtros se etiquetan como FILTER. Para el curso (datos de prueba) se usó un filtro de cobertura de 10 lecturas, lo cual fue considerado mínimo para poder determinar de forma adecuada el genotipo, pero es arbitrario.
Combinar vcfs filtrados
Juntamos los archivos vcf anotados de SNPs e Indels en uno solo Comando:
java -jar GenomeAnalysisTK.jar \
-T CombineVariants \
-R reference.fasta \
--variant:foo muestra_FILTERED_SNP.vcf \
--variant:bar muestra_FILTERED_INDEL.vcf \
-o muestra_FILTER_VARIANTS.vcf \
-genotypeMergeOptions PRIORITIZE \
-priority foo,bar
Ejemplo:
java -jar /opt/GenomeAnalysisTK-3.7-0/GenomeAnalysisTK.jar -T CombineVariants -R /datos/reference/genomes/hg19_reference/hg19.fasta --variant:foo S10_FILTERED_SNP.vcf --variant:bar S10_FILTERED_INDEL.vcf -o S10_FILTER_VARIANTS.vcf -genotypeMergeOptions PRIORITIZE -priority foo,bar
Tarea
- Seguir este tutorial con los datos de la muestra previamente elegida. Todas las muestras son de pacientes, para los cuales se sospechaba de una mutación patogénica. Se realizó una secuenciación de un panel de genes con equipamiento MiSeq.
- En materiales y métodos del reporte, indique el número de genes incluidos en el panel e incluya una tabla con la lista de genes (consejo: revise el archivo regiones_blanco.bed). Indique también la región genómica total (en pares de bases) cubierta por el panel, o sea, el tamaño de las regiones blanco (consejo: revise su reporte qualimapReport.html).
- Realice el filtrado de variantes con dos filtros, DP<10 y uno adicional que usted proponga.
- Estime cuántas variantes son eliminadas por el filtro DP<10 solamente, y cuántas por ambos filtros.
- Genere un reporte e incluya una tabla con el número de variantes detectadas totales, SNPs, e INDELs. Para cada caso, indicar el número de variantes filtradas y que pasaron los filtros (solo uno, y ambos)
- Visualice una variante en IGV, mostrando tracks tanto para el alineamiento (bam) como las variantes detectadas (VCF).
- Asegúrese de usar un tamaño de ventana que muestre suficiente detalle como para leer la secuencia de referencia, pero sin un zoom excesivo para que se logre ver algo de contexto de secuencia. Ojalá que se vean otras variantes al rededor de la central. Incluya un track con los genes. Si no se ve ningún gen cercano a la variante, elija otra variante.
- En resultados, indique en formato de tabla el número de variantes detectadas según ubicación (intrónica, río arriba, río abajo, codificante con cambio de sentido, sin sentido, etc).
- Realice una anotación de las variantes con la herramienta en línea VEP. Asegúrese de usar la versión del genoma que utilizó en el alineamiento. Incluya anotaciones de Significancia clínica y puntajes CADD. Baje la tabla de variantes anotadas en formato TXT y fíltrela (por ejemplo en R) para generar una tabla que solo contenga variantes con un valor distinto a “benign” en la columna “CLIN_SIG” o un valor de CAAD > 20. Incluya incluya la tabla filtrada en su informe (si hubo variantes que pasaron los filtros) e interprete sus resultados.
- En la sección conclusiones, asegúrese de concluir algo sobre la muestra (presencia o no de mutaciones con potencial patogénico).