BioinfinvRepro
Curso de introducción a la bioinformática e investigación reproducible
Unidad 3 Introducción a R con un enfoque bioinformático
R y RStudio
¿Qué es R?
- R es un lenguaje de programación y un ambiente de cómputo estadístico
- R es software libre (no dice qué puedes o no hacer con el software), de código abierto (todo el código de R se puede inspeccionar - y se inspecciona).
- Cuando instalamos R, instala la base de R. Mucha de la funcionalidad adicional está en paquetes que la comunidad crea.
¿Por qué R?
- R funciona en casi todas las plataformas (Mac, Windows, Linux).
- R es un lenguaje de programación completo, permite desarrollo de DSLs (funciones muy específicas)
- R promueve la investigación reproducible: no sólo de análisis sino de cómo se hicieron las figuras.
- R está actualizado gracias a que tiene una activa comunidad. Solo en CRAN hay cerca de 8000 paquetes (funcionalidad adicional de R creadas creada por la comunidad).
- R se puede combinar con herramientas bioinformáticas y formatos de datos procesados (e.g. plink, vcf, etc) para realizar análisis y figuras.
- R tiene capacidades gráficas muy sofisticadas.
- R es popular como herramienta estadística (la guerra del software) y cada vez más también como herramienta bioinformática.
- Además del repositorio de paquetes de todo tipo de R (CRAN) también hay un repositorio especializado en paquetes de bioinformática (Bioconductor)
Descargar R y RStudio
Para comenzar se debe descargar R, esta descarga incluye R básico y un editor de textos para escribir código. Después de descargar R se recomienda descargar RStudio (gratis y libre).
RStudio es un ambiente de desarrollo integrado para R: incluye una consola, un editor de texto y un conjunto de herramientas para administrar el espacio de trabajo cuando se utiliza R.
¿Cómo entender R?
- Hay una sesión de R corriendo. La consola de R es la interfaz entre R y nosotros.
- En la sesión hay objetos. Todo en R es un objeto: vectores, tablas, funciones, etc.
- Operamos aplicando funciones a los objetos y creando nuevos objetos.
La consola (o terminal)
Es una ventana que nos permite comunicarnos al motor de R. Esta ventana acepta comandos en el lenguaje de R y brinda una respuesta (resultado) a dichos comandos.
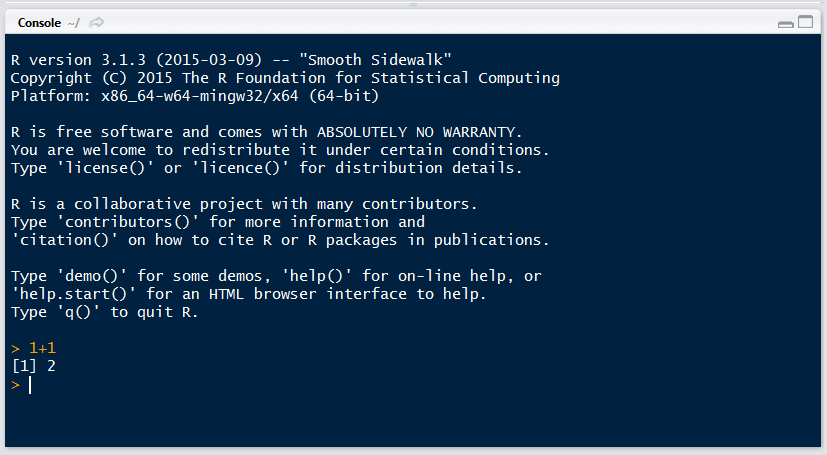
Por ejemplo, le podemos pedir a R que sume 1+1 así:
1+1
La consola se distingue por tener el símbolo > seguido de un cursor parpadeante que espera a que le demos instrucciones (cuando recién abrimos R además muestra la versión que tenemos instalada y otra info).
Tu Consola debe verse más o menos así después del ejemplo anterior:
Scripts y el editor
Como hemos visto en otras unidades, un script es un archivo de nuestros análisis que es:
- un archivo de texto plano
- permanente,
- repetible,
- anotado y
- compartible
En otras palabras, un script es una recopilación por escrito de las instrucciones que queremos enviar a la consola, de modo que al tener esas instrucciones cualquiera pueda repetir el análisis tal cual se hizo.
Un script muy sencillo podría verse así:
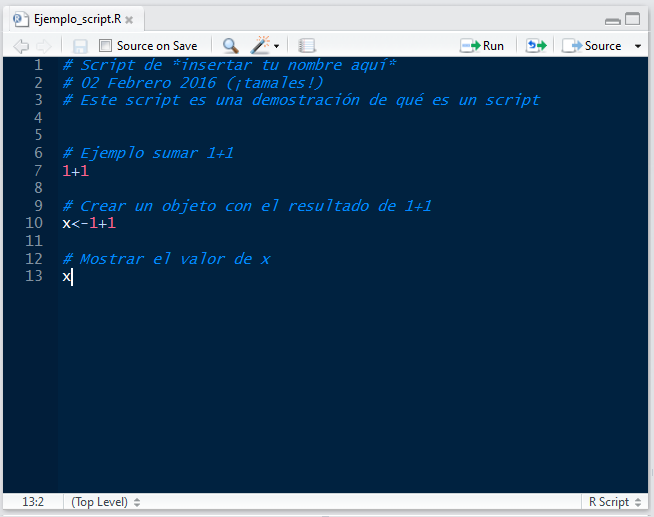
“Ejemplo_script.R” es el nombre del archivo, es decir, que este texto es un archivo de texto (con terminación .R en vez de .txt) que vive en mi computadora.
RStudio brinda además de la consola un editor de texto, que es la pantalla que se observa en el ejemplo anterior. Lo que escribas en el editor de texto puede “enviarse” a la consola con los shortcuts de abajo o con el ícono correr.
La idea es que en el editor de texto vayas escribiendo los comandos y comentarios de tu script hasta que haga exactamente lo que quieras.
Algunos shortcuts útiles en RStudio:
En el editor
- command/ctrl + enter: enviar código a la consola
- ctrl + 2: mover el cursor a la consola
En la consola
- flecha hacia arriba: recuperar comandos pasados
- ctrl + flecha hacia arriba: búsqueda en los comandos
- ctrl + 1: mover el cursor al editor
- ? + nombre de función: ayuda sobre esa función.
Funciones básicas de R más importantes para bioinformática
R Básico
Imprime dos veces este Acordeón de R básico. Ten uno siempre contigo y otro bajo la almuada para la ósmosis.
Antes de pasar a las funciones bionformáticas, veamos la sintaxis básica de R y los principales comandos que aprender.
En resumen:
- Expresiones matemáticas:
1+1 - Strings de texto:
"¡Holaaaaa mundo!" - Valores lógicos:
1<5,2+2 ==5 -
Crear una variable u objeto:
x<-5 - Funciones: son un comando que hace algo específico dentro de R.
A diferencia de bash, en R los comandos, o funciones, tienen la sintaxis: “nombre_de_la_funcion()”. Entre los paréntesis van los argumentos de la función, es decir los datos o parámetros con los cuales queremos que la función haga algo. Para ver qué hace una función, podemos correr en la terminal ?nombre_de_la_funcion, lo cual abrirá la ayuda detallando los argumentos y cualquier otra información e cómo usar dicha función.
Ejemplo de funciones: sum(), mean() , log().
Vamos a ver l ayuda de la función log() con ?log.

Ejercicio: crea una variable con el logaritmo base 10 de 50 y súmalo a otra variable cuyo valor sea igual a 5.
Ahora vamos a ver con detalle el resto de objetos de R en estas notas sobre los tipos de objetos de R básico. Abajo te dejamos un resumen:
Vectores:
- vectores
c(5, 4, 6, 7),5:9 - Acceso a elementos de un vector
[]
Ejercicio: suma el número 2 a todos los números entre 1 y 150.
Ejercicio ¿cuántos números son mayores a 20 en el vector -13432:234?
Matrices
- Matrices
matrix(0, 3, 5) - Acceso a elementos e una matriz
[ , ]
Data frames
- Data frame
data.frame(x = c("a", "b", "c"), y = 1:3) - Acceso a elementos e una data.frame
[ , ],$
Cargar archivos:
read.delim sirve para cargar un archivo de texto con filas y columnas. Revisa su ayuda para determinar qué parámetros utilizar para leerlo si está separado por comas, tabulaciones (tab), espacios o qué.
Además de archivos de filas y columnas, se pueden leer a R todo tipo de archivos, en algunos casos esto se hace con paquetes que crearon funciones específicas para esto. Normalmente se llaman read.algo. Por ejemplo la función read.plink del paquete snpMatrix.
Cuando utilices read.delim o símil, asume que tu WD es donde vive tu script y utiliza rutas relativas para navegar hasta el archivo que deseas cargar.
(Para poner triste Alicia preguntar por qué es importante hacer lo anterior).
Working directory
Buena práctica recomendada: que tu working directory sea donde sea que viva el script en el que estás trabajando.
Para averiguar cuál es tu WD actual utiliza getwd().
Puedes definir tu WD manualmente con la función setwd(), pero OJO: realiza esto en La Consola, NO en tu script. Neto, porfas.
Una trampa práctica en RStudio para que tu WD sea el lugar donde vive tu script es ir al Menú:
Session > Set Working Directory > To source file location
O sease “source file” = tu script activo.
Nota también que si abres RStudio clickeando su ícono, tu WD por default será el home de tu usuario. Sin embargo, si abres RStudio clickeando en un script, el WD default será donde viva dicho script.
Ejercicio:
Carga en R el archivo Prac_Uni3/maices/meta/maizteocintle_SNP50k_meta_extended.txt y ponlo en un objeto de R llamado meta_maiz.
Trabajar con paquetes y datos externos
R base contiene las funciones más básicas, pero la verdadera riqueza de R está en sus paquetes. Estos los puede desarrolar cualquier persona y publicar. Van desde análisis estadísticos generales hasta funciones muy específicas pera determinado campo. En CRAN puedes explorar la gran gama de paquetes sobre cualquier tema. Algunos paquetes son especializados para bioinformática, como adegenet y ape. Puedes ver una lista de más paquetes relacionados con genética estadística en CRAN Task Statistical Genetics.
Otra opción para encontrar paquetes útiles es googlear “R package” + keywords de tu tema de interés, por ejemplo “metabarcoding”.
(También hay un repositorio especializado en paquetes de bioinformática (Bioconductor), que veremos más adelante)
install.packages sirve para instalar un paquete en nuestras máquinas, esto la baja de CRAN u otro servidor y lo instala en nuestro R, pero no lo carga a la sesión activa. Este paso hay que hacerlo solo una vez.
Una vez que el paquete está instalado, este NO estará cargado en el cerebro de R al menos que utilicemos library(nombredelpaquete). Este paso hay que hacerlo cada vez que corras R para un análisis que ocupe dichos paquetes.
Si tu script utiliza un paquete determinado, es recomendable que estos se carguen en las primeras líneas o al principio de la sección de código que los utilizará. Sólo carga los paquetes que realmente utilices en un script dado.
Cómo citar R
Citar R:
citation("base")
Citar un paquete en particular:
citation("NombrePaquete")
(o lo que loas autoreas especifiquen en su sitio web)
Acordeón funciones útiles al trabajar con paquetes y archivos de datos
- Funciones de sistema:
list.files,getwd,setwd - Cargar una función:
source - Instalar paquetes (sola una vez en cada equipo):
install.packages. - Cargar un paquete previamente instalado (cada vez que corramos el script):
library. - Cargar a R un archivo de texto con filas y columnas (separado por tabs o comas):
read.delim. - “Pegar” texto uno detrás de otro:
paste()ypaste0().
For loops
Al igual que en bash, en R pueden hacerse for loops, con la siguiente sintaxis:
for (counter in vector) {commands}
Ejemplo:
for (i in 2:10){
print(paste(i, "elefantes se columpiaban sobre la tela de una araña"))
}
La anterior es la versión más simple de un loop. Para otras opciones (como while, if, if else, next) revisa este tutorial.
Los loops son útiles ya que nos permiten reciclar código en vez de repetir lo mismo para difernetes valores. Por ejemplo el loop anterior hace lo mismo que:
paste(2, "elefantes se columpiaban sobre la tela de una araña")
paste(3, "elefantes se columpiaban sobre la tela de una araña")
paste(4, "elefantes se columpiaban sobre la tela de una araña")
paste(5, "elefantes se columpiaban sobre la tela de una araña")
paste(6, "elefantes se columpiaban sobre la tela de una araña")
paste(7, "elefantes se columpiaban sobre la tela de una araña")
paste(8, "elefantes se columpiaban sobre la tela de una araña")
paste(9, "elefantes se columpiaban sobre la tela de una araña")
paste(10, "elefantes se columpiaban sobre la tela de una araña")
Ejercicio
-
Escribe un for loop para que divida 35 entre 1:10 e imprima el resultado en la consola.
-
Modifica el loop anterior para que haga las divisiones solo para los números nones (con un comando, NO con
c(1,3,...)). Pista:next. -
Modifica el loop anterior para que los resultados de correr todo el loop se guarden en una df de dos columnas, la primera debe tener el texto “resultado para x” (donde x es cada uno de los elementos del loop) y la segunda el resultado correspondiente a cada elemento del loop. Pista: el primer paso es crear un vector fuera del loop. Ejemplo:
elefantes<-character(0)
for (i in 2:10){
elefantes<-rbind(elefantes, (paste(i, "elefantes se columpiaban sobre la tela de una araña")))
}
elefantes
Ejercicio
Abre en RStudio el script Prac_Uni3/mantel/bin/1.IBR_testing.r. Este script realiza un análisis de aislamiento por resistencia con Fst calculadas con ddRAD en Berberis alpina.
Lee el código del script y determina:
- ¿qué hacen los dos for loops del script?
- ¿qué paquetes necesitas para correr el script?
- ¿qué archivos necesitas para correr el script?
Funciones propias: crear funciones y utilizarlas con source
source es una función que sirve para correr un script de R dentro de otro script de R. Esto permite modularizar un análisis y luego correr una pipeline general, así como tener por separado funciones propias (que podemos llamar igual que llamamos las funciones de los paquetes) y que utilizamos mucho en diversos scripts. Este tipo de funciones son las que podemos compartir en Github con otros usuarios y hasta convertirlas en un paquete.
Ejemplos de cómo utilizar source: correr el script del ejercicio anterior desde otro script con la línea.
source("1.IBR_testing.r")
Nota que pare que esto funcione tu working directory debe ser el correcto para leer 1.IBR_testing.r como si fuera un archivo (que lo es). Es decir tu WD debe ser la ruta donde está 1.IBR_testing.r (Unidad3/PracUni3/mantel/bin/1.IBR_testing.r)
Hacer una función propia:
Este es el esqueleto de una función escrita dentro de R:
myfunction <- function(arg1, arg2, ... ){
statements
return(object)
}
Ojo: el comando return es necesario al final de una función siempre que queramos que dicha función “devuelva” un objeto (por ejemplo una df que creemos como parte de la función). De no poner esta instrucción, la función correrá desde otro script, pero no veremos ningún resultado.
Ejemplo:
give_i_line<- function(file, i){
## Arguments
# file = path to desired file with the indicadores, must be tab delimited and do NOT have a header
# i = number of line of file we want to print
## Function
# read indicadores list
indicador<-read.delim(file, header=FALSE, quote="", stringsAsFactors=FALSE)
# give text of the i line of the file
x<-indicador[i,1]
return(x)
}
give_i_line("../data/indicadores.txt", i=2)
x<-give_i_line("../data/indicadores.txt", i=2)
Como alternativa a return() puedes poner el nombre del objeto (como si quisieras verlo en la terminal).
give_i_line<- function(file, i){
## Arguments
# file = path to desired file with the indicadores, must be tab delimited and do NOT have a header
# i = number of line of file we want to print
## Function
# read indicadores list
indicador<-read.delim(file, header=FALSE, quote="", stringsAsFactors=FALSE)
# give text of the i line of the file
x<-indicador[i,1]
x
}
give_i_line("../data/indicadores.txt", i=2)
x<-give_i_line("../data/indicadores.txt", i=2)
Si quieres ver un resultado pero que este no sea guardado como un objeto, utiliza print().
give_i_line<- function(file, i){
## Arguments
# file = path to desired file with the indicadores, must be tab delimited and do NOT have a header
# i = number of line of file we want to print
## Function
# read indicadores list
indicador<-read.delim(file, header=FALSE, quote="", stringsAsFactors=FALSE)
print(i)
# give text of the i line of the file
x<-indicador[i,1]
x
}
give_i_line("../data/indicadores.txt", i=2)
x<-give_i_line("../data/indicadores.txt", i=2)
Si guardamos la función como un script llamado give_i_line.r después podemos correrla desde otro script, llamándola con source():
source("give_i_line.r")
give_i_line("../data/indicadores.txt"), i=2)
Nota que source NO corre la función en sí, sino que solo la carga al cerebro de R para que podamos usarla como a una función cualquiera de un paquete.
El nombre del archivo R no improta, pero es buena práctica ponerle el mismo que el nombre de la función.
Ejercicio: Escribe una función llamada calc.tetha que te permita calcular tetha dados Ne y u como argumentos. Recuerda que tetha =4Neu.
Ejercicio: Al script del ejercicio de las pruebas de Mantel, agrega el código necesario para realizar un Partial Mantel test entre la matriz Fst, y las matrices del presente y el LGM, parcializando la matriz flat. Necesitarás el paquete vegan.
Rmarkdown y R Notebook
R Markdown es un formato que te permite crear documentos o reportes, en los que al mismo tiempo guardas y ejecutas código.
Primero, instala R Markdown:
install.packages("rmarkdown")
Ahora puedes crear un archivo .Rmd en “Archivo > Nuevo archivo > R Markdown”. (nota que estoy es distinto que R script).
Un archivo R Markdown es un archivo de texto plano que debe verse algo así:
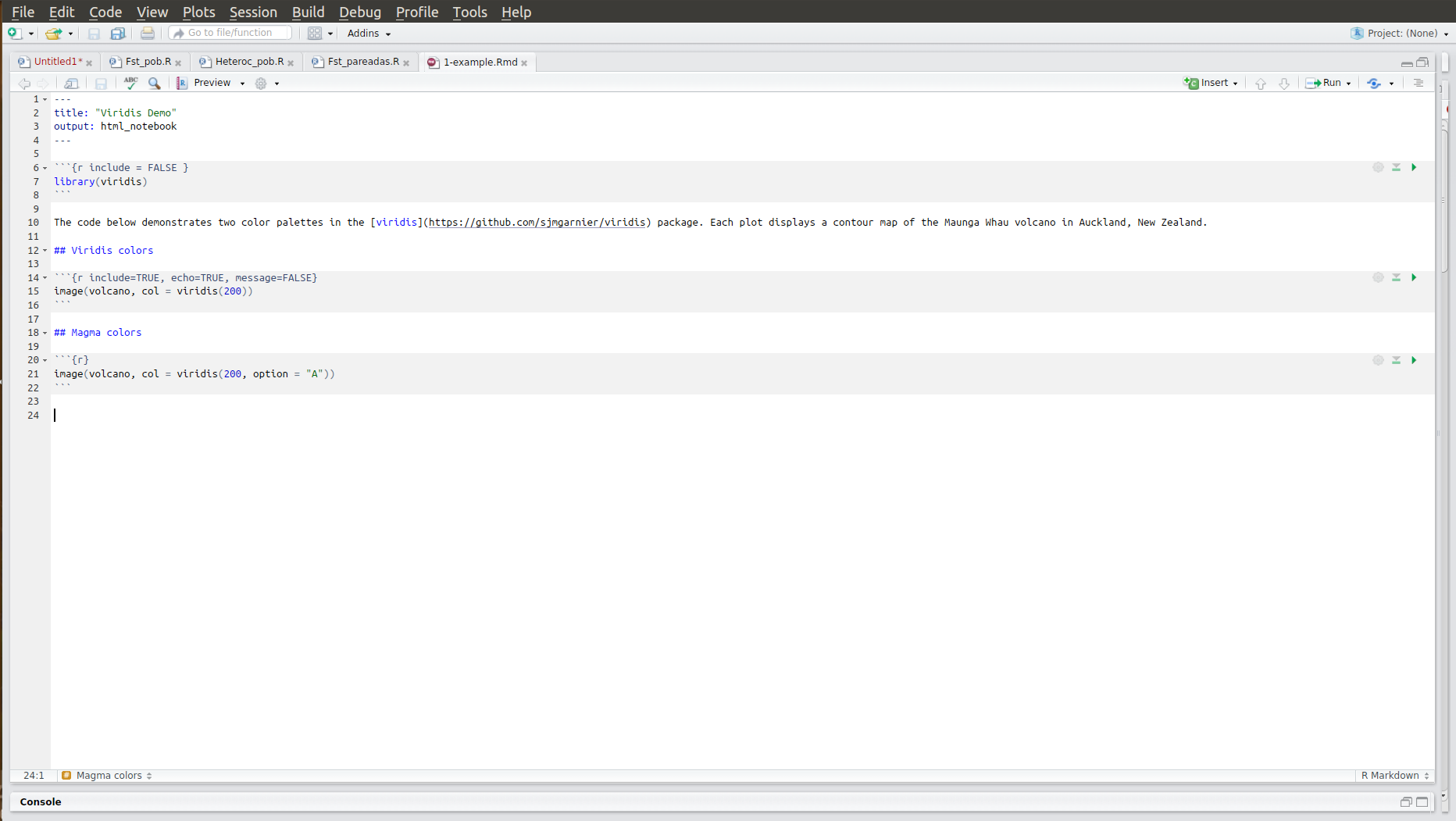
El archivo tiene tres tipos de contenido:
- Encabezado (—)
- Código (```)
- Texto simple (Markdown)
Notarás que se pueden ejecutar las líneas de código de forma independiente e interactiva, y que el output (lo que saldría en la consola o los plots) del código de muestra debajo de éste.
Cuando abres o creas un archivo Rmd la interfaz de RStudio cambia. Ahora, puedes ejecutar el código usando las flechas y los resultados se despliegan a continuación del código.
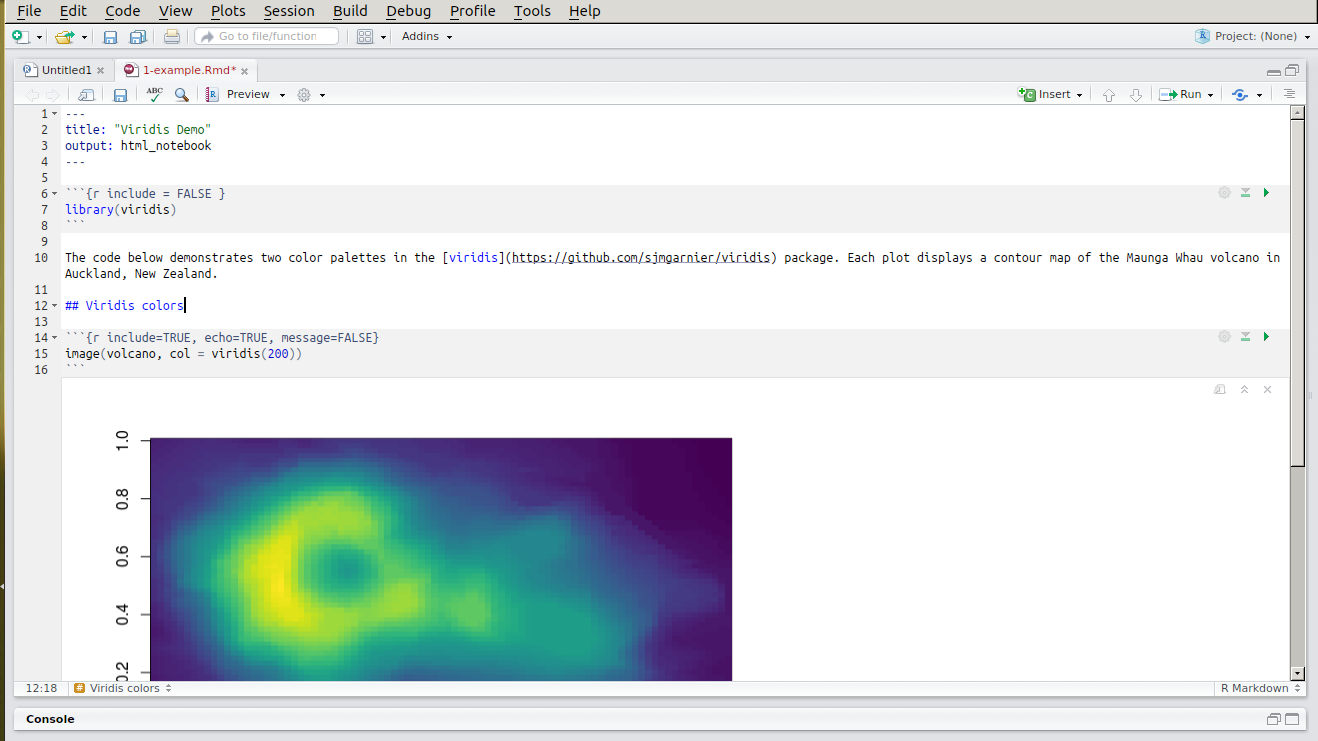
Archivos de salida
A partir de un archivo .Rmd, es posible crear archivos de salida en una gran variedad de formatos, por ejemplo:
- HTML
- Documentos interactivos
- Word
- Diapositivas
- Páginas web
- R Notebooks
Checa más formatos de salida aquí
Para crear el reporte o archivo de salida, debes correr render(), dar click en “Knit” o ⇧+Ctrl+K.
Cuando se renderea un archivo .Rmd, también se crea un archivo html. Este es un archivo HTML que contiene el código y los output resultantes, que puede abrirse en cualquier explorador web y en RStudio.
Generar un reporte de un script .R
Es posible generar un reporte a partir de un script de R, aún cuando no tenga el formato de un archivo R Markdown. Únicamente das click en el cuaderno (Compile report) o Ctrl+Shift+K.
¿Rmd o reporte de script.R?
Un Rmarkdown es más útil si quieres hacer un reporte de los análisis, o incluso escribir tu tesis, utilizando R, de forma que tanto el texto explicativo como el código estén integrados en el mismo archivo. Esto garantiza la reproducibilidad total, pero solo funciona si todo lo vas a hacer con R y si te acomoda que otros comenten tu documento así.
Un reporte de un script.R funciona si quieres “guardar cómo se ve” correr un análisis o discutirlo con tus colegas, pero no son un reporte tan bonito para entregar a alguien externo.
Así que cuándo usar cuál depende de la situación y tus gustos.
Manipulación y limpieza de datos en R
dplyr es un paquete super útil para hacer operaciones de transformación de datos. Vamos a ver su Data Transformation Cheat Sheet para aprender sus comandos principales.
Puedes ver más acordenos parecidos en este link
La manipulación y la limpieza da datos muchas veces es necesaria antes de poner hacer análisis en R. Aquí trataremos brevemente lo siguientes puntos en [notas aparte]:
Ejercicio:
Escribe un script que debe estar guardado en Prac_Uni3/maices/bin y llamarse ExplorandoMaiz.R, que 1) cargue en R el archivo Prac_Uni3/maices/meta/maizteocintle_SNP50k_meta_extended.txt y 2) responda lo siguiente.
(averigua cada punto con comandos de R. Recuerda comentar o tendrás 7 años de mala suerte en el lab)
-
¿Qué tipo de objeto creamos al cargar la base?
-
¿Cómo se ven las primeras 6 líneas del archivo?
-
¿Cuántas muestras hay?
-
¿De cuántos estados se tienen muestras?
-
¿Cuántas muestras fueron colectadas antes de 1980?
-
¿Cuántas muestras hay de cada raza?
-
En promedio ¿a qué altitud fueron colectadas las muestras?
-
¿Y a qué altitud máxima y mínima fueron colectadas?
-
Crea una nueva df de datos sólo con las muestras de la raza Olotillo
-
Crea una nueva df de datos sólo con las muestras de la raza Reventador, Jala y Ancho
-
Escribe la matriz anterior a un archivo llamado “submat.cvs” en /meta.
Graficar en R
Los apuntes de esta sección están en Graficar en R (código y gráficas) y Graficar en R (sólo código)
Bioconductor
Como hemos visto los paquetes son un grupo de funciones que alguien desarrolla en torno a un tema específico. Muchos paquetes viven en CRAN. Pero también hay otros repositorios más especializados. Bioconductor es un repositorio de paquetes de R especializaos en en análisis de datos genómicos y de secuenciación masiva. Es decir, en Bioconductor encontrarás paquetes que NO están en CRAN.
![]()
Generalidades de Bioconductor
Página principal de Bioconductor
Paquetes de Bioconductor
Como los paquetes de Bioconductor están escritos en el lenguaje de R, muchos tendrán tipos de objetos particulares al paquete y funciones nuevas, pero con tener las bases de R que hemos visto estarás listoa para aprenderlo.
La mejor manera de conocer qué hace y usar un paquete es seguir un tutorial o vignette.
Por ejemplo esta de ggtree y esta de SNPRelate.
Además, BioConductor desarrolló una clase de objetos, llamados GRanges que permite almacenar y manipular intervalos genómicos y variables definidas a lo largo de un genoma. Los GRanges son particularmente útiles para representar y analizar anotaciones genómicas y alineamientos y son la base de varios paquetes de Bioconductor
Los GRanges están definidos en el paquete GenomicRanges. Este tutorial explica su funcionamiento básico y lo recomiendo mucho. También está bueno el Capítulo 9 Working with Range Data del libro de Vince Buffalo Bioinformatics Data Skills.
Workflows
Para algunas tareas comunes en análisis genéticos, como Variant calling.
Cursos y conferencias de Bioconductor
Hay muchos cursos y conferencias sobre Bioconductor que ocurren anualmente y que van de temas generales a especializados en algún proceso. Para una intro general yo recomiendo el curso online Bioconductor for Genomic Data Science de Kasper D. Hansen que incluye videos y código con notas en R y html.
En el 2018, por primera vez en Latinamérica, se dió un curso de nivel intermedio-avanzado: Latin American R/BioConductor Developers Workshop 2018. Estén atentxs para nuevas ediciones.
Instalar Bioconductor y sus paquetes
1) Tener instalado R
2) Instalar bioconductor (source al script biocLite.R que nos permitirá instalar paquetes de Bioconductor).
source("https://bioconductor.org/biocLite.R")
biocLite()
(Si lo anterior manda algún error intenta http:// en vez de https://)
3) Utilizar la función biocLite para instalar los paquetes deseados. Ejemplo:
biocLite("ggtree")
Nota: algunos paquetes necesitan pasos extra de instalación, como jalar algo de GitHub, pero esto será indicado en la documentación del paquete.
Cómo citar R y Bioconductor
Citar R:
citation("base")
Citar Bioconductor:
citation("Biobase")
Citar un paquete en particular:
citation("NombrePaquete")
(o lo que loas autoreas especifiquen en su sitio web)
Ejercicio: divídanse por equipos de 2-3 personas según su tipo de datos o análisis. Exploren los paquetes de CRAN y de Bioconductor. Compartan por el gitter el link a la página de descripción del paquete y mencionen brevemente por qué les parece útil.
Los 25 pasos que no debes olvidar siempre que trabajes en R.
-
En tu computadora crea un directorio para tu proyecto. Aquí pondras los datos y los scripts. Se recomienda que los datos estén en un subdirectorio llamado
datay los scripts en uno llamadobinoscripts. -
Guarda tus datos en el directorio
data. Si los tienes en Excel guarda una versión en formato de texto (separado por comas o por tabulaciones). -
Has un back up de tus datos en algún lugar diferente a tu computadora. OSF es un buen lugar.
-
Crea un repositorio de github en el directorio donde están guardados tus scripts.
-
Abre R y crea tu script. Guárdalo en
bin(o donde hayas decidido guardar tus scripts). -
Escribe a manera de comentarios cuál es el objetivo de tu script y cuáles son los principales pasos que crees debes hacer (algoritmo).
-
Desde bash (o conectando RStudio con Github), has un commit de tu trabajo hasta ahora (
git addy luegogit commit -"create script to do ....") -
Toma un descanso.
-
Cambia tu wd al lugar donde guardaste tu script. Para hacer esto NO escribas
setwd()en tu script. Mejor, utilizasetwd()en la Consola o, si no quieres escribir la ruta a tu script, ve al menú “Session > Set Working directory > To source file location”. -
Al inicio de tu script carga con
library()los paquetes que utilizará tu script. Si no los tienes instalados, instálalos coninstall.packages(), corriendo dicho comando en la consola pero no escribiéndolo en los comandos de tu script. -
Después de cargar paquetes, pon una sección donde cargues los datos. Esto puedes hacerlo con funciones de la familia
read.comoread.delim()oread.csv. Para esto recuerda:- dar al argumento
filela ruta relativa desde donde está guardado tu script hasta donde están guardado tu archivo de datos. Por ejemplofile="../data/mis_datos.txt"si estás usando la estructura de este ejemplo. - revisa los argumentos de tu función read para asegurarte que los datos se carguen como quieres, por ejemplo
header = TRUEsi la primera fila de tus datos tiene el nombre de las columnas. - crea un objeto (variable) con el archvio que estás leyendo, es decir con
<-. Por ejemplo:
- dar al argumento
my_df <- read.delim(file="../data/mis_datos.txt")
-
Revisa que tus datos sean tus datos y se vean como tus datos. Para esto: - Ve tu data frame dando click en el objeto que creaste (aparece en el panel de “Enviroment” de RStudio. O con el comando
View(my_df). Este comando lo puedes correr en la consola, pero no lo pongas en tu script. - Utilizanrow()yncol()para ver que tus datos tengan el número de filas y columnas que esperas. - Revisa los levels (ejemplolevels(my_df$genero)) de las variables categóricas de tus datos, esto sirve para identificar typos. - Revisa, revisa, revisa. -
Asegúrate de que cada paso esté bien comentado hasta ahora.
-
git commit -m "load data" -
Come algo.
-
Explora tus datos con gráficas
-
git commit -m "add exploratory plots" -
Asegúrate que esté bien comentado tu código.
-
Haz pruebas estadísticas para comprobar tus hipótesis. Apóyate de esta tabla:

REF: Bekcerman et al (2012). Getting Started with R: An Introduction for Biologists.
-
git commit -m "add stats analyses" -
Si creaste funciones y las usas frecuentemente, ponlas en un script separado y cárgalas al inicio de tu script con
source(). -
Asegúrate que tu script corra de principio a fin. Para esto borra el cerebro de R con
rm(list=ls())y vuelve a correr tu script desde el principio. -
Comunica tus resultados en un reporte html. Recuerda que puedes crearlo con el botón
knitsi tu script es un .Rmd, o con el botón “Compile report” si tu script es un script de R. -
git commit -m "knit report" -
Revisa tu código, especilamente los comentarios. Asume que no vas a volver a tocar tu script en 6 meses. ¿Te faltó algo? ¿Puedes escribir comentarios para tu yo del futuro que digan qué te faltó y por donde ir?
-
Ve por cervezas o a socializar de algún otro modo. Cuéntale al mundo lo bonito que es R (pero elige sabiamente cuándo detenerte).
Primeros auxilios cuando algo no te corre en R
- Te falta una
, - Te sobra un espacio
- Cierra tus
() - El texto va entre
" " - Si no pusiste
<-no has creado el objeto - No estás donde crees que estás (encuéntrate con
getwd())