BioinfinvRepro
Curso de introducción a la bioinformática e investigación reproducible
Análisis clustering
Ricardo Verdugo y Karen Oróstica
Datos
Para este tutorial, utilizaremos la matriz datos normalizados que generamos en el tutorial Análisis de expresión diferencial en R
En el tutorial pasado, generamos un objeto normdata. Vaya la carpeta DE_tutorial, vuelva a importar los datos y normalizarlos. Una vez generada la matrix de datos, expórtela como un archivo plano separado por tabulaciones.
outdir <- "output"
if(!file.exists(outdir)) {
dir.create(outdir, mode = "0755", recursive=T)
}
Data.Raw <- read.delim("Illum_data_sample.txt")
signal <- grep("AVG_Signal", colnames(Data.Raw)) # vector de columnas con datos
detection <- grep("Detection.Pval", colnames(Data.Raw))
annot <- read.delim("MouseRef-8_annot.txt")
probe_qc <- ifelse(annot$ProbeQuality %in% c("Bad", "No match"), "Bad probes",
"Good probes")
design <- read.csv("YChrom_design.csv")
print(design)
Data.Raw <- Data.Raw[probe_qc %in% "Good probes",]
annot <- annot[probe_qc %in% "Good probes",]
rawdata <- as.matrix(Data.Raw[,signal])
rownames(rawdata) <- Data.Raw$PROBE_ID
colnames(rawdata) <- design$Sample_Name
library(preprocessCore)
normdata <- normalize.quantiles(rawdata)
colnames(normdata) <- colnames(rawdata)
rownames(normdata) <- rownames(rawdata)
probe_present <- Data.Raw[,detection] < 0.04
detected_per_group <- t(apply(probe_present, 1, tapply, design$Group, sum))
present <- apply(detected_per_group >= 2, 1, any)
normdata <- normdata[present,]
annot <- annot[present, ]
write.table(normdata, file.path(outdir, "normdata.txt"), sep="\t", row.names=T)
Luego cree una nueva carpeta para este tutorial e inicie una sesión de R usado esa capeta como directorio de trabajo.
Tutorial
Este tutorial está basado en el Cluster Analysis, Quick-R, DataCamp.
Importe los datos
mydata <- read.delim("../output/normdata.txt", as.is=T)
Cambie los nombres de las columnas para que sea más fácil identificar el grupo experimental en los siguientes gráficos.
design <- read.csv("../YChrom_design.csv")
colnames(mydata) <- design$Group
Preparar los datos
mydata <- na.omit(mydata) # eliminación filas con faltantes
mydata <- scale(mydata) # estandarizar variables
Particionamiento (Clustering)
K-means clustering es el método de partición más popular. Requiere que el analista especifique la cantidad de clusters que desea extraer. Una gráfica de la suma de cuadrados dentro de los grupos por número de grupos extraídos puede ayudar a determinar el número apropiado de grupos. El analista busca una curva en la gráfica similar a una prueba de evaluación en el análisis factorial. Ver Everitt y Hothorn (pág. 251). Es decir, queremos el menor k que maxima la disminución de la suma de cuadrados. Una vez que la suma de cuadrados se estabiliza, no hay beneficio en seguir aumentado el k.
# Determinar el número de grupos
wss <- (nrow(mydata)-1)*sum(apply(mydata,2,var))
for (i in 2:15) wss[i] <- sum(kmeans(mydata,
centers=i)$withinss)
png("SSQ_by_K_using_kmeans.png", width=600, heigh=500)
plot(1:15, wss, type="b", xlab="Number of Clusters",
ylab="Within groups sum of squares")
dev.off()
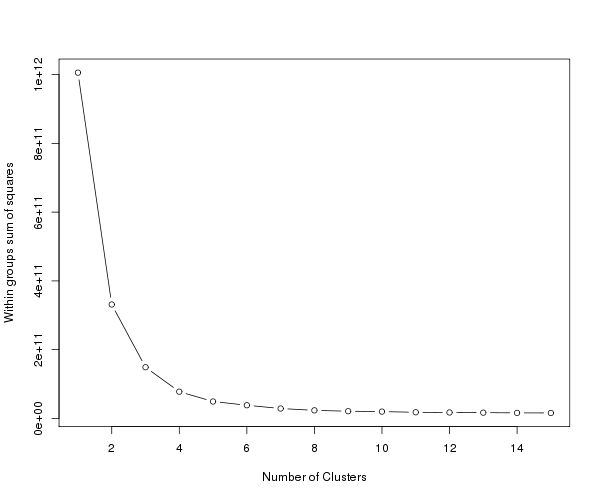
En base a este gráfico, un K=5 parece un número razonable de clusters, así que usaremos ese valor.
fit <- kmeans(mydata, 5) # 5 cluster solution
class(fit)
# [1] "kmeans"
# append cluster assignment
mydata2 <- data.frame(mydata, fit$cluster)
head(mydata2)
B.I BY.I B.C BY.C B.C.1 B.I.1
ILMN_1248788 207.3817 250.1129 221.9986 252.7182 246.3669 201.0584
ILMN_2896528 10069.3537 9936.6241 9839.0757 12435.8400 11339.6023 10165.8597
ILMN_2721178 2146.1743 2037.4831 2084.2946 2705.3736 2556.7989 2216.5564
ILMN_1227723 1138.9989 1089.6837 927.3898 927.3898 1274.3487 1081.4380
ILMN_3033922 1855.7668 1797.0263 1855.7668 1799.6424 2099.7710 1891.1571
ILMN_3092673 8047.4719 7507.8776 7601.7383 7321.8261 8452.9129 8237.6955
BY.I.1 BY.C.1 BY.C.2 B.C.2 BY.I.2 B.I.2
ILMN_1248788 267.1375 214.2343 199.2973 219.7224 236.7965 194.6496
ILMN_2896528 8948.2579 12255.4739 11542.1811 10615.6144 10615.6144 9511.4862
ILMN_2721178 2155.8708 2497.0567 2713.5130 2166.2265 2072.5239 2022.4722
ILMN_1227723 1151.0470 924.1201 1107.6753 1202.7625 1136.6648 1050.2442
ILMN_3033922 1771.0536 1870.0569 1791.2293 1911.4861 1722.9694 1836.4394
ILMN_3092673 6723.2857 7943.6365 8237.6955 8452.9129 7601.7383 8150.2006
B.I.3 BY.I.3 BY.C.3 B.C.3 fit.cluster
ILMN_1248788 211.8343 207.8115 221.9986 171.3909 4
ILMN_2896528 10028.7244 10885.4851 12435.8400 9029.3847 2
ILMN_2721178 2078.4187 2354.4808 2724.6773 2376.6771 1
ILMN_1227723 1172.4656 1149.4365 976.4341 1114.9443 4
ILMN_3033922 1891.1571 1859.8280 1949.1789 1739.1569 4
ILMN_3092673 7479.2309 8047.4719 8237.6955 7479.2309 2
¿Haría algún procesamiento adicional a los datos de expresión para que el clustering sea óptimo? ¿Podría la distribución de los afectar las estimación de promedios, los que son importantes para los métodos paramétricos como k-means? Si este fuera el caso, ¿qué podría realizar para solucionarlo?
Calcule los promedios de expresión por cluster
aggregate(mydata,by=list(fit$cluster),FUN=mean)
Group.1 B.I BY.I B.C BY.C B.C B.I
1 1 52412.040 52412.0399 52412.0399 52412.0399 52412.0399 52412.0399
2 2 3732.378 3733.8785 3725.5083 3727.6995 3723.2772 3730.3724
3 3 23674.468 23774.2967 23506.7469 23412.2511 23435.3683 23368.0363
4 4 10291.461 10256.0496 10344.5888 10355.3966 10336.0652 10399.2822
5 5 528.928 528.7892 529.9703 530.5617 531.2235 528.7974
BY.I BY.C BY.C B.C BY.I B.I B.I
1 52412.0399 52412.0399 52412.0399 52412.040 52077.8121 52077.8121 52412.040
2 3735.4716 3732.0670 3726.4979 3723.584 3731.5504 3739.0103 3736.976
3 23716.5112 23451.7654 23647.4590 23725.561 23951.5896 23516.6848 23774.297
4 10250.9351 10337.8046 10296.6688 10268.616 10226.1668 10345.5624 10242.462
5 529.3597 530.1607 530.0147 530.410 530.3385 530.0446 528.868
BY.I BY.C B.C
1 51966.727 52412.0399 52412.040
2 3731.080 3729.4663 3724.861
3 23836.102 23673.2230 23735.708
4 10278.669 10282.1466 10272.214
5 530.441 529.8188 529.758
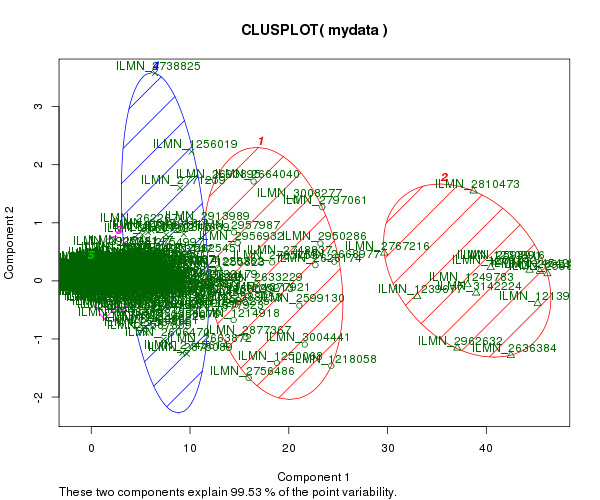
Gráfico de clústers contra los 2 primeros componentes principales de varianza
library(cluster)
png("Clusplot_kmeans_k5.png", width=600, heigh=500)
clusplot(mydata, fit$cluster, color=TRUE, shade=TRUE,
labels=2, lines=0)
dev.off()
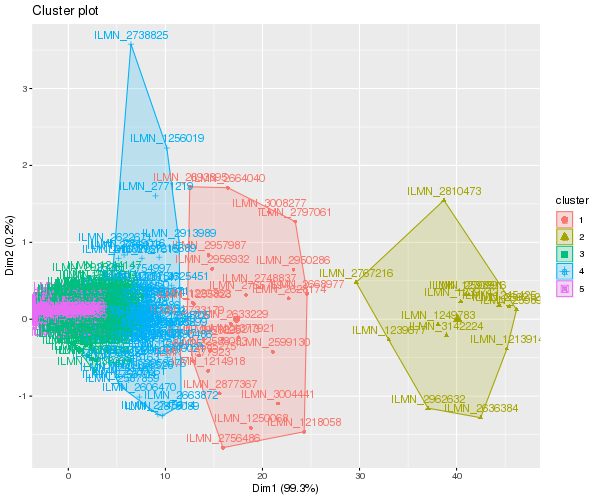
Utilizar la librería factoextra que genera gráficcos basados en ggplot. La función fviz_cluster() genera gráficos para métodos basados en particionamiento, incluyendo kmeans, pam, Mclust, entre otros.
library(factoextra)
png("fviz_cluster_kmeans_k5.png", width=600, heigh=500)
fviz_cluster(fit, data = mydata)
dev.off()
Se puede invocar una versión robusta de K-means basada en mediods usando pam () en lugar de kmeans(). La función pamk() en el paquete fpc es una envoltura para pam que también imprime el número sugerido de grupos en función del ancho de silueta promedio óptimo.
Clúster jerárquico
Hay una amplia gama de enfoques de agrupamiento jerárquico. He tenido buena suerte con el método de Ward que se describe a continuación.
d <- dist(t(mydata), method = "euclidean") # distance matrix
fit <- hclust(d, method="ward.D")
png("hclust_samples.png", width=600, heigh=500)
plot(fit, hang = -1, cex = 0.8) # display dendogram
dev.off()
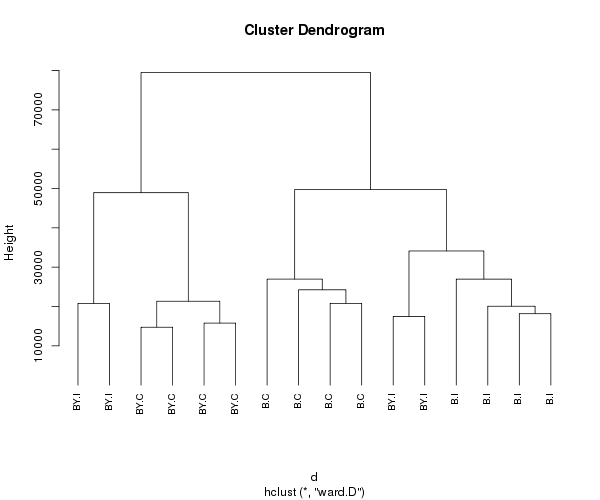
Visualice los groups
groups <- cutree(fit, k=5) # cut tree into 5 clusters
groups
# B.I BY.I B.C BY.C B.C B.I BY.I BY.C BY.C B.C BY.I B.I B.I BY.I BY.C B.C
# 1 2 3 4 3 1 2 4 4 3 5 1 1 5 4 3
Dibuje cuadrados al rededor de los clusters cuando el arbol se corta para generar 5 clusters
png("rect_hclust_samples.png", width=600, heigh=500)
plot(fit, hang = -1, cex = 0.8) # display dendogram
rect.hclust(fit, k=5, border="red")
dev.off()
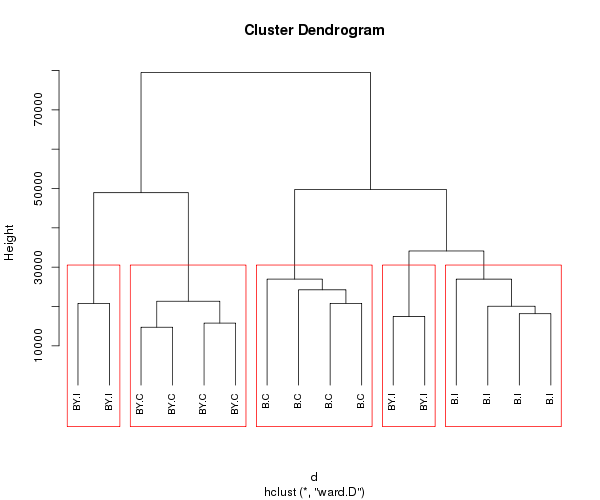
Clústering jerárquico de genes
Usaremos la función get_dist() del paquete factoextra, que a diferencia de la función dist(), puede calcular distancias en base a la correlación de pearson. Esta medida es preferible cuando no nos interesa agrupar genes por su nivel promedio de expresión, sino por su patrón de expresión.
gd <- get_dist(mydata, method ="pearson", stand=FALSE)
gfit <- hclust(gd, method="ward.D")
png("hclust_genes.png", width=600, heigh=500)
plot(gfit, hang = -1, cex = 0.8, labels = FALSE) # display dendogram
dev.off()
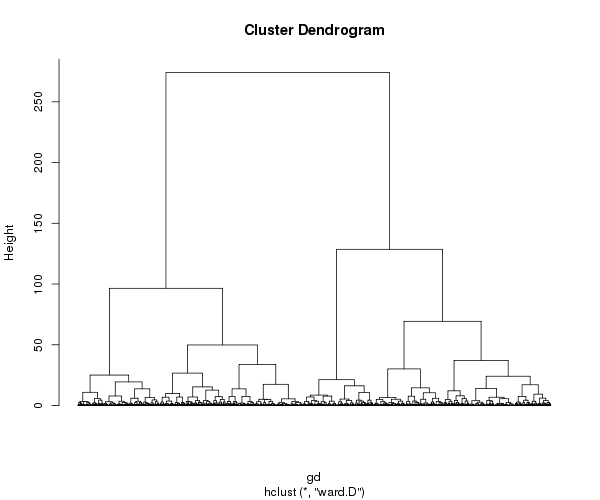
Tarea
Usando solo los genes seleccionados por expresión diferencial obtenidos en el tutorial de expresión diferencial, ya sea por tratamiento, genotipo o por interacción:
- Realice un particionamiento jerárquico de sus muestras con la medida de distancia euclidiana
- Realice un particionamiento jerárquico de sus sondas usando el complemento de la correlación de Pearson como la medida de distancia.
- Asegúrese de realizar los procesamientos de datos necesarios para que la distribución y escala de los datos sean apropiadas para la metodología de clústeres utilizada en cada caso.
- Genere gráficos de suma de cuadrados para sondas y para muestras
- Basándose en los gráficos de sumas de cuadrados, elija el k más apropiado en su criterio para sondas y para muestras
- Agregue rectángulos a los particionamientos jerárquicos (nota: en su informe puede mostrar solo el árbol final, con los rectángulos).
- Guarde su trabajo como un informe en formato pdf.