BioinfinvRepro
Curso de introducción a la bioinformática e investigación reproducible
Unidad 4 Introducción al software especializado
Lecturas recomendadas sobre software especializado y reproducibilidad:
Utilidad de software especializado
El software especializado permite realizar operaciones complejas con datos genéticos y preguntas biológicas de un tipo particular.
Por ejemplo el ensamblado de novo de datos de GBS/RAD, puede hacerse con diferente software dependiendo de si las preguntas biológicas están enfocadas en genética de poblaciones (e.g. Stacks), filogenética (e.g. pyRAD) o diversidad de cultivos (e.g. UNEAK-TASSEL).
En la siguiente parte del curso veremos diferentes sofwares para cada tipo de datos, pero no profundizaremos mucho en cada uno. Recuerda que ese no es el objetivo del curso y para esto hay cursos especializados. La idea es que puedas entender los artículos y el manual de cualquier software para tomar tus propias decisiones y realizar tus análisis con cualquier software.
Documentación de software especializado
La documentación de un programa bioinformático (y de cualquier software) se refiere al manual y tutoriales asociados al programa.
Dos maneras de encontrar el manual:
- Dentro del programa (e.g.
man vcftoolsovcftools -help) - En internet (en la página del software).
La complejidad del manual depende de la complejidad de la herramienta en sí. Si es algo que involucre varios pasos y analizar (en vez de solo e.g. reformatear) datos lo más recomendable es dedicarle una tarde (o semana) entera a correr y entender paso por paso un tutorial. Otras cosas sencillas pueden entenderse dándole un vistazo al manual.
Vamos a ver algunos ejemplos de manuales online:
Instalación manual
El software especializado en bioinformática en su mayoría corre directamente desde la línea de comando Unix y está escrito en lenguajes como C, python y Perl, entre otros. Por ende, su instalación no es dar dos clicks, sino realizar una serie de pasos, en su mayoría involucrando los siguientes pasos:
1) Busca la versión más reciente del software especializado de tu interés. Por lo general tienen su propia página en algún rincón del internet.
Veamos dos ejemplos:
2) Busca la sección de Download, o Install. Normalmente te llevarán a bajar un tar o a un comando para bajar el tar desde Github.
Ojo: si hay una sección que diga “Computing Requirements” o “Specs” checa eso primero. E.g: para Trinity.
3). Baja el source code del programa.
Si vemos el contenido de los tar que bajamos veremos algo así:
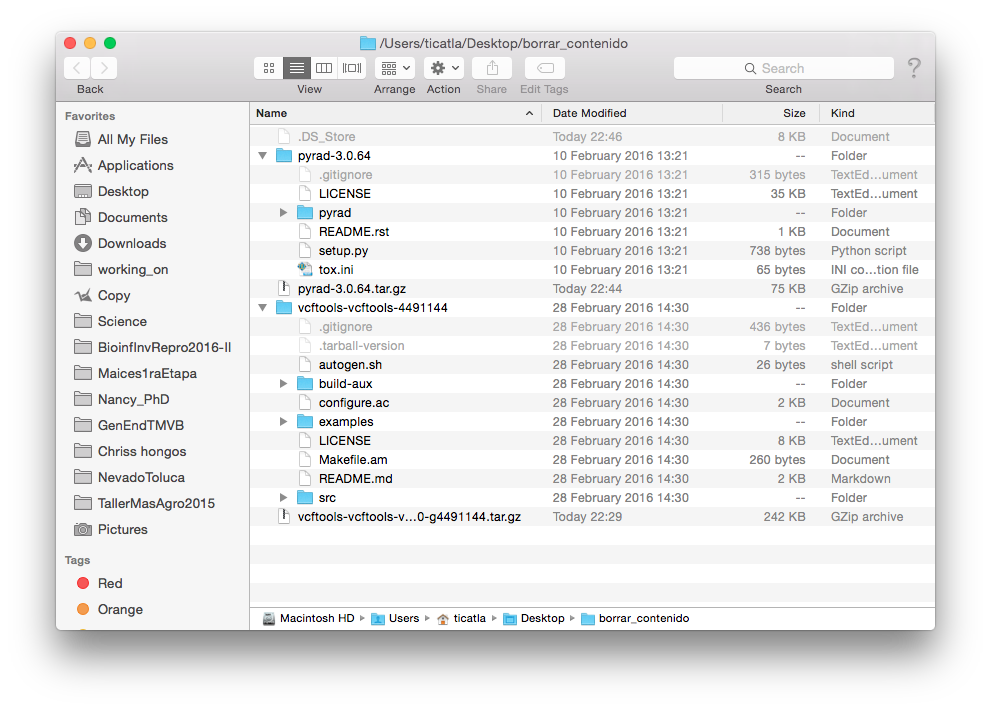
- Por lo general el tar con el source code incluye un archivo de texto (o Markdown) llamado README, pero también puede llamarse INSTALL. Este es el primer archivo que siempre debemos consultar (y si tanto README como INSTALL existen, primero README).
Así se ve el de pyRAD:
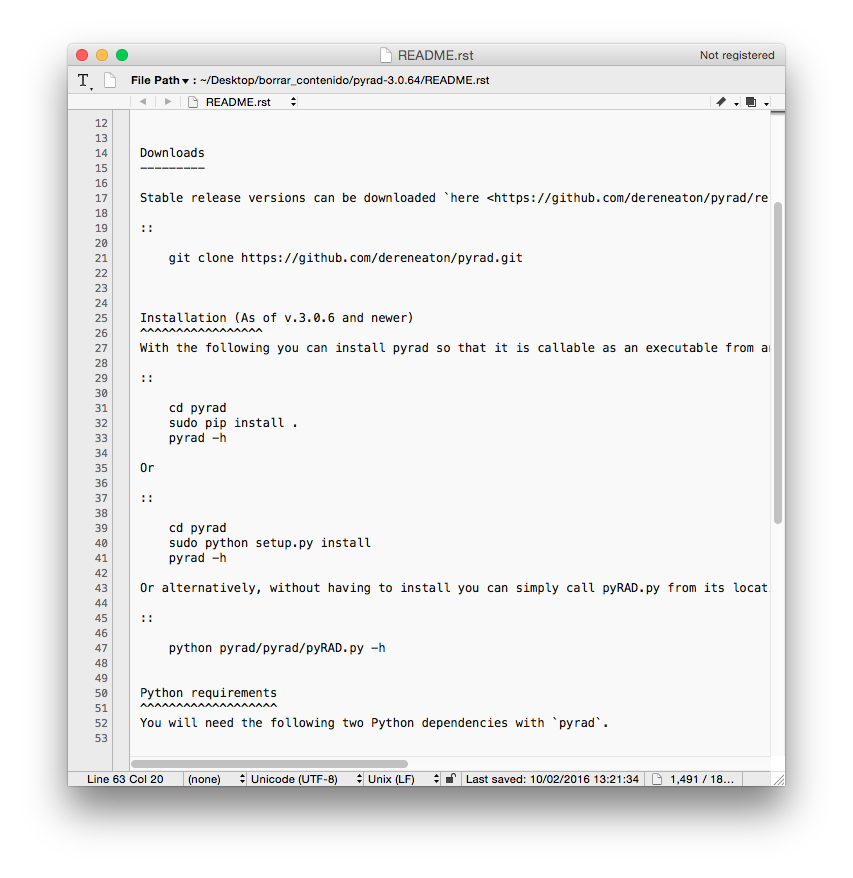
Y así el de VCFtools:
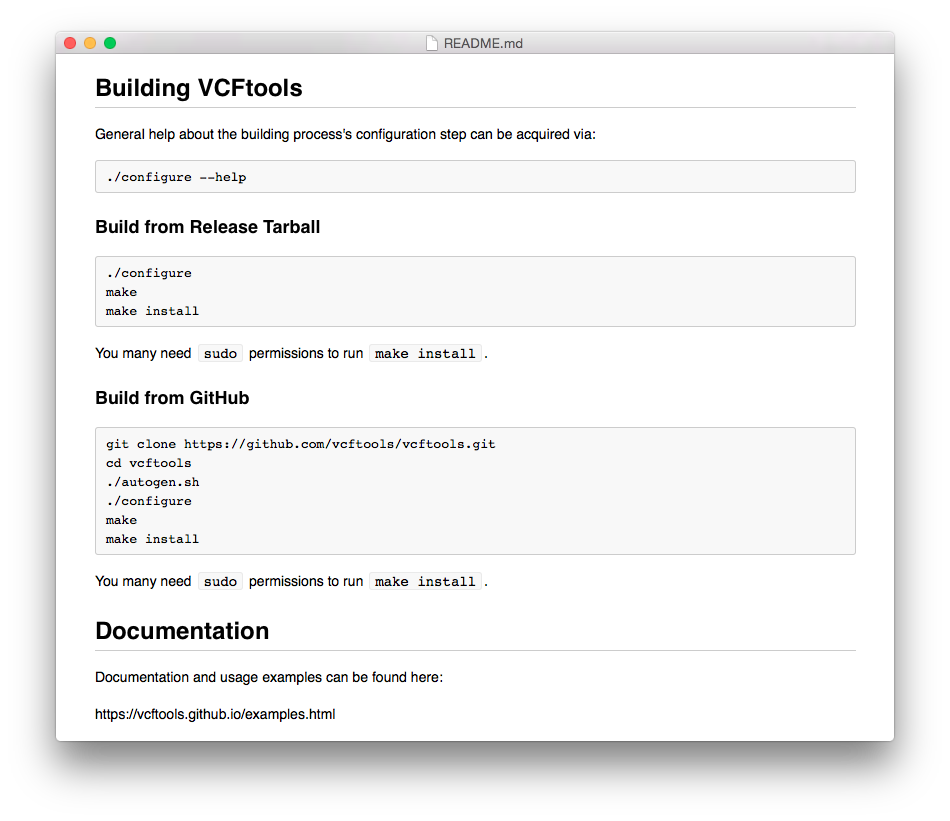
- La instalación consiste en seguir los pasos que nos dice el README (o el link que contenga el README, a veces pasa).
Puntos a observar:
- La instalación varía de programa a programa
- La instalación puede requerir los comandos:
sudo./configuremakemake install
- Nuestro software de interés puede requerir dependencias, es decir otros programas o librerías (de python, Perl, etc) para poder correr.
- Algunas de esos otros programas pueden instalarse a través de programas para instalar cosas, como
pip,brew, etc.
¿Qué hacen sudo, ./configure, make y make install?
Bonita referencia aquí.
sudo:
Te vuelve el super usuario, es decir eres un todo poderoso administrador que puede hacer lo que sea con la línea de comando sin que la computadora te diga que no.
xkcd lo explica así:

Debemos usar sudo ya que al correr como usuarios normales no tenemos privilegios de administrador, lo cual es una medida de seguridad para que no corran programas que se metan con nuestro sistema sin nuestra autorización, y también para revisar que sabes lo que haces antes de modificar algo importante en el sistema.
./configure:
Para correr esto primero debes estar en el tar (descomprimido) del programa que acabas de bajar. La razón es que esta línea de comando corre un script llamado configure (que es un ejecutable) que viene con tu programa.
Un script configure básicamente revisa que la computadora donde vas a instalar el programa tenga las dependencias que el programa necesita. En ocasiones puede decirte que no las tiene, pero que amablemente las baja por ti. En tal caso te lo preguntará, con al final de la pregunta un paréntesis así (Y/n), tienes que decirle si Yes or Not.
Si algunos de los requisitos importantes no se puede conseguir configure marcará error y no podrás continuar con la instalación hasta que tengas esas dependencias en orden (o sea que las instales por ti misma). Este suele ser el paso más doloroso de instalaciones difíciles.
Si todo sale bien configure habrá creado un nuevo archivo en nuestro WD (que sigue siendo el descomprimido del tar del programa en cuestión) llamado MakeFile, puedes pasar al siguiente paso:
make:
make es un programa de Unix que corre un archivo MakeFile que se encuentre en el WD. Este archivo (que creamos con configure en el paso anterior) es ligeramente diferente dependiendo de cada computadora, es decir, está personalizado para tu computadora, por eso no viene en el tar que bajamos.
El MakeFile indica la secuencia de comandos que se necesitan seguir para construir (build) los componentes o sub-programas del software y crea los ejecutables necesarios. A esto también se le conoce como compilación.
Si todo sale bien, puedes seguir al último paso:
make install:
make install también corre el archivo MakeFile, pero sólo una sección dentro de dicho archivo llamada Install. Esta sección contiene instrucciones para que los archivos creados por el paso anterior (make) se copien a sus directorios destino dentro de tu computadora, por ejemplo /usr/local/bin para que cualquier usuario los pueda correr. Si fue necesario instalar dependencias, estas también se copiarán al lugar que les corresponda (se crearon en el paso anterior, pero vivían en un directorio temporal).
Muy probablemente en este paso tu terminal te dirá que no tienes autorización (Permission denied) si intentas correr make install. Aquí es cuando entra sudo, así:
sudo make install
(Te pedirá tu pasword, dáselo).
Ejercicio: encuentra la documentación de un programa que se utilice con tu tipo de datos. Contesta:
- ¿La documentación explica la instalación, si sí, cuáles son los pasos?
- ¿Puedes encontrar de un vistazo lo que te interesa saber cómo hacer?
- ¿Cuenta con un tutorial?
- ¿Cuánto tiempo crees te tomará correr el tutorial?
Trabajar en servidores remotos
Muchos análisis bioinformáticos, especialmente si involucran datos genómicos, requieren de muchas horas de procesamiento o de muchos recursos computacionales (memoria RAM y espacio de disco).
Ejemplos de recursos computacionales necesarios para:
- Ensamblaje de novo genoma entero: RAM RAM RAM (>256 GB). Tarda 1 semana a 4 meses (según sp.)
- Ensamblaje de novo datos GBS/RAD: RAM (4-16 GB). Horas a días, pero se hacen varias pruebas.
- Mapeo a genoma referencia: RAM (8 GB ok). Horas a días
- Análisis filogenéticos bayesianos: proco RAM, pero pueden duarar semanas.
- Análisis de estructura poblacional: poco RAM, varias horas.
Invertir en estos recursos vale más la pena si se comparten entre varios usuarios, por lo que muchas veces las instituciones tienen un cluster de super cómputo. Un cluster es un conjunto de computadoras (procesadores) conectadas entre sí a las que nos conectamos y asignamos trabajos para que corran entre varios procesadores. Los clusters operan con algún sabor de Linux, por lo que nuestra interacción con el es vía la línea de comando.
Normalmente el cluster físicamente existe en algún lugar al que no tenemos acceso físico, por lo que más bien funciona como un servidor al que nos conectamos.
Conectarse a un servidor
Para conectarnos a un servidor primero tenemos que contactar a su admin y tener un nombre de usuario, contraseña y probablemente claves de seguridad. Ya que tenemos esto, desde una terminal y teniendo acceso a internet podemos conectarnos con shh:
ssh viene de “secure shell”, permite conectarnos via nuestra consola de shell a un servidor que tenga un puerto abierto al internet. Entonces, en so forma más sencilla, tenemos que decirle nuestro nombre de usuario y en qué servidor, como si fuera una dirección de correo:
ssh usuario@servidor
Pero si el servidor tiene un sistema de seguridad que requiera que tengas llaves privadas (un archivo que vive en tu computadora y que solo tu tienes y que el servidor conoce), entonces será algo así:
ssh -v -i <ruta-a-tu-llave-privada> usuario@servidor
Luego te pedirá tu password y listo.
- Nota: quizá la primera vez que hagas el
shha un servidor te diga algo como esto:
The authenticity of host 'node07.conabio.gob.mx (172.16.15.29)' can't be established.
ECDSA key fingerprint is SHA256:NsiFXj.......
Are you sure you want to continue connecting (yes/no)?
Dile que yes.
Al hacer esto “entraremos” al servidor y podemos crear archivos y correr scripts desde nuestra terminal. Nota que tu tu terminal cambiará y dejarás de ser tu en tu computadora y pasarás a ser el usuario con el que hiciste log in en el servidor. Por ejemplo en el servidor de CONABIO que ocuparemos seré: [cirio@node07 ~]$
Para cerrar tu sesión debes correr el comando exit. Volverás a ser tu, en tu computadora.
Ejercicio Preparamos un servidor de CONABIO al que podremos entrar durante la clase. Entra al servidor con el shh que te indicaremos en el chat y google classroom. Luego crea un directorio que sea igual a tu nombre de usuario de github o la primera letra de tu nombre y tu primer apellido. Copia el directorio BioinfinvRepro (que ya existe) de home a tu nuevo directorio.
Correr scripts
Podemos trabajar de forma interactiva (“en vivo” igual que en nuestra compu), pero si te sales se mueren tus trabajos. Para dejar scripts corriendo y podernos salir hay dos formas:
-
Correr scripts en el background (REF). Por ejemplo con
nohup script &. -
Utilizar un sistema de colas, como SLURM (o el que use tu cluster). Este sistema permite que varios usuarios corran sus cosas al mismo tiempo, pero pero asignando recursos (por ejemplo número de procesadores, cantidad de RAM) independientes a cada trabajo. Así, si no hay recursos disponibles en lugar de que el cluster colapse, simplemente se espera a terminar de correr un proceso antes de continuar con los otros.
Con squeue podemos ver cómo está la cola. Y para especificar cuántos recursos consumirá nuestro script se agregan un par de líneas iniciando con #SBATCH (parecen comentarios) después del she-bang al inicio de nuestro script. Estos detalles deben venir en el sitio de documentación del cluster que ocupes, por ejemplo el del nodo genómico de CONABIO, se ve así:
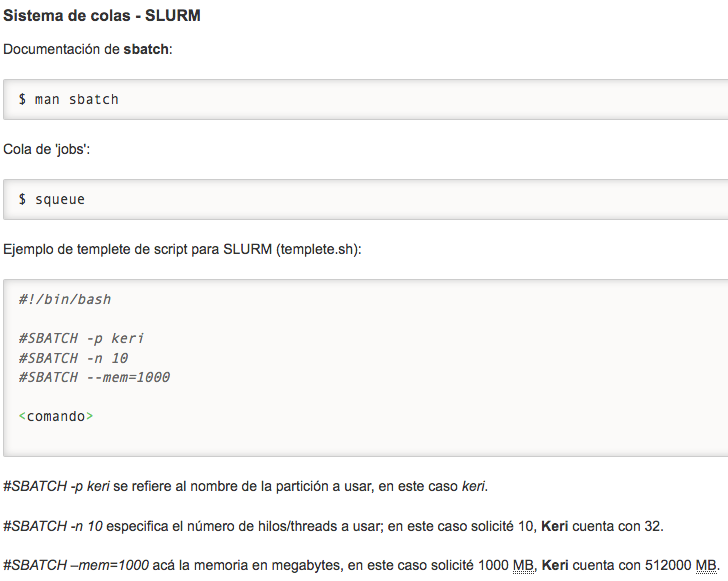
De forma que un script ejemplo queda así:
#!/bin/sh
# #SBATCH -p keri
# #SBATCH -n 2
# #SBATCH --mem=32000
# define variable for taxa name
taxa=LuxuNica
## Run admixture
# mamke output to put results
mkdir -p ../data/genetic/admixture_outs/$taxa
### Run admixutre
for K in {1..7};
# run admixture
do ./admixture --cv ../data/genetic/filtered/bytaxa_${taxa}.bed $K | tee ../data/genetic/admixture_outs/${taxa}/log${K}.out;
# move output to output folder
mv bytaxa_${taxa}.$K* ../data/genetic/admixture_outs/$taxa
done
# move output
# save CV in a single file
#grep -h CV ../data/genetic/admixture_outs/${taxa}/log*.out > ../data/genetic/admixture_outs/${taxa}/Kerror_${taxa}.txt
Luego para correr nuestro script, se hace con el comando sbatch myscript.sh.
Siempre es buena idea ver cómo anda nuestro cluster de trabajo con htop.
Transferir archivos
- El comando
scp:
scp ArchivoAcopiarLocal usuario@servidor:/RutaDondeCopiar
- Sofware como Filezilla.
El problema de las versiones y OS para la reproducibilidad
A veces no basta con tener los scripts, tener una versión distinta de un software puede llevar a resultados diferentes.
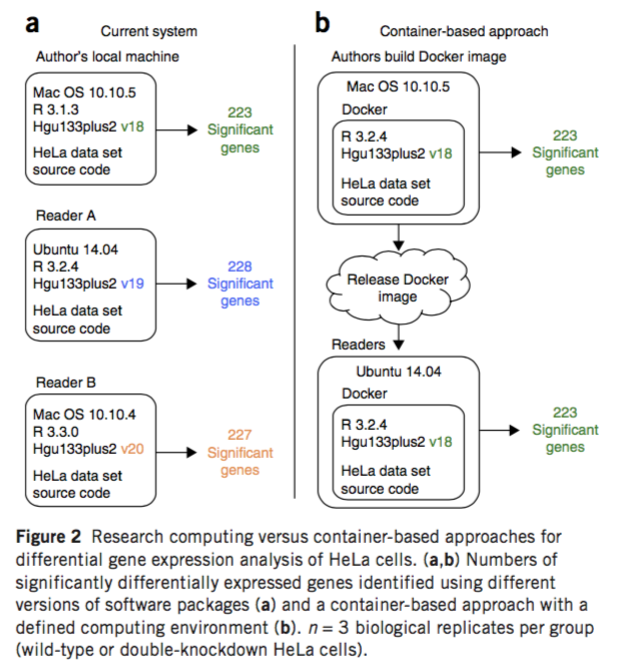
Figura tomada de Beaulieu-Jones et al Greene (2017)
Las pipelines bioinformáticas ocupan muchos programas distintos que constantemente están renovándose. La probabilidad de que tengamos una versión de algo distinta a la que corrieron los autores originales es muy alta.
Como vimos arriba, instalar un software especializado puede no ser trivial, y además al instalar un programa bioinformático es común “romper” las dependencias de otro programa o incluso tu sistema operativo. Por eso a la hora de tener que hacer una actualización nos dan mido los tiburones:

Docker
Una solución a ambos problemas es utilizar un sistema de contenedores de sofware, como Docker.
_logo.png”)
Docker permite poner dentro de un contenedor un software (o varios) junto con todo lo que necesitan para correr: su sistema de archivos, código, herramientas del sistema, librerías, etc, cualquier cosa que normalmente podamos instalarle a un sistema operativo.
Terminología básica:
-
Un contenedor es una versión de Linux reducida a sus componentes más básicos.
-
Una imagen es el software que cargamos en un contenedor.
-
Un dockerfile es un script que describe (e instala) el software que pondremos en una imagen, pero esto no incluye sólo el programa en sí, sino también cualquier detalle de la configuración del ambiente y hasta los comandos que queremos corra.
Esto nos permite que un programa corra de manera idéntica sin importar el sistema operativo original del equipo, y hace que la instalación sea independiente de la instalación de otro software.
Los contenedores son la nueva forma de hacer aplicaciones.
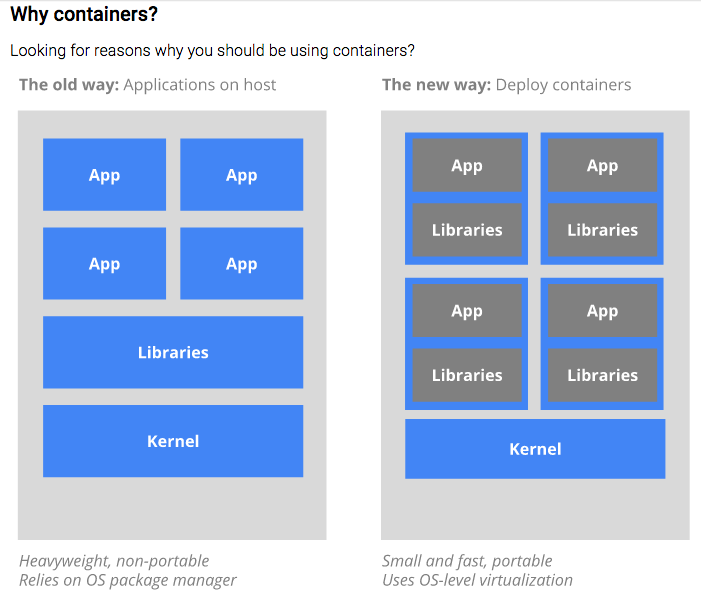
Figura tomada de la documentación de Kubernetes, que es otra forma de hacer contenedores. Su documentación tiene una muy buena intro a por qué utilizar contendores.
En el siguiente link puedes encontrar las instrucciones y primeros pasos para utilizar Docker.
Para probar que Docker está funcionando correctamente deberás correr docker run hello-world y obtener:
$ docker run hello-world
Unable to find image 'hello-world:latest' locally
latest: Pulling from library/hello-world
78445dd45222: Pull complete
Digest: sha256:c5515758d4c5e1e838e9cd307f6c6a0d620b5e07e6f927b07d05f6d12a1ac8d7
Status: Downloaded newer image for hello-world:latest
Hello from Docker!
This message shows that your installation appears to be working correctly.
To generate this message, Docker took the following steps:
1. The Docker client contacted the Docker daemon.
2. The Docker daemon pulled the "hello-world" image from the Docker Hub.
3. The Docker daemon created a new container from that image which runs the
executable that produces the output you are currently reading.
4. The Docker daemon streamed that output to the Docker client, which sent it
to your terminal.
To try something more ambitious, you can run an Ubuntu container with:
$ docker run -it ubuntu bash
Share images, automate workflows, and more with a free Docker ID:
https://cloud.docker.com/
For more examples and ideas, visit:
https://docs.docker.com/engine/userguide/
Cómo iniciar Docker:
Para inciar Docker primero prendemos la máquina (para poder correr los comandos de docker, esto es similar a prender una máquina virtual en VirtualBox).
Puede ser que la máquina ya esté prendida, para averiguarlo puedes correr un comando como:
docker ps o (en Linux sobretodo) sudo docker ps (te pedirá tu contraseña).
Si obtienes algo parecido a esto todo está en orden.
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
Si obtienes un error como “Cannot connect to the Docker daemon. Is the docker daemon running on this host?” Entonces deberás iniciar la máquina:
-
En Mac: Click el ícono de Docker. Dependiendo de tu sistema esto abrirá una terminal con docker corriendo o prenderá docker (probablemente lo verás en el menú cerca de donde está el wireless de tu compu) de manera que ya podrás utilizar
docker OPTIONSdesde tu terminal. -
En Linux:
sudo service docker start
Para no tener que utilizar sudo cada vez que corres un comando de docker para no tener que prenderlo cada vez que reuinicias tu compu, no olvides revisar este link
Funcionamiento básico de Docker
La sintaxis de docker es: docker OPTIONS. En Linux probablemente tendrás que correrlo con sudo antes, al menos que hagas lo que dice . Así: sudo docker OPTIONS. Te pedirá tu password (el que usas para entrar a tu computadora).
Las opciones más importanes de Docker son:
pulluna imagen (solo la primera vez)runla imagen dentro de un contenedor (para crearlo, solo la primera vez)exitpara salir del contendorstoppara detener un contenedorrestartpara reactivar un contenedorexecpara entrar a un contenedor activormborrar un contenedor (debesstopprimero).rmiborrar una imagen.
Veámoslo con un ejemplo:
1) Bajar (pullear) una imagen con pull. Por ejemplo la última versión de ubuntu:
$ docker pull ubuntu #Baja la última versión de ubuntu
Using default tag: latest
latest: Pulling from library/ubuntu
5a132a7e7af1: Pull complete
fd2731e4c50c: Pull complete
28a2f68d1120: Pull complete
a3ed95caeb02: Pull complete
Digest: sha256:4e85ebe01d056b43955250bbac22bdb8734271122e3c78d21e55ee235fc6802d
Status: Downloaded newer image for ubuntu:latest
Aquí por default bajó la última, pero también hubieramos podido especificar qué versión de ubuntu queríamos, así: docker pull ubuntu:14.04
Para revisar hayamos bajado la imagen deseada:
$ docker images #Enlista imagenes ya bajadas
REPOSITORY TAG IMAGE ID CREATED SIZE
ubuntu latest 07c86167cdc4 11 days ago 188 MB
hello-world latest 690ed74de00f 5 months ago 960 B
docker/whalesay latest 6b362a9f73eb 9 months ago 247 MB
2) Cargamos la imagen dentro de un contenedor con run. Voilá, estamos dentro de un Ubuntu, específicamente dentro de un contenedor corriendo Ubuntu.
$ docker run -it ubuntu bash
root@8f36b66384cb:/#
root@8f36b66384cb:/# ls
bin dev home lib64 mnt proc run srv tmp var
boot etc lib media opt root sbin sys usr
root@8f36b66384cb:/# mkdir Prueba
root@8f36b66384cb:/# ls
Prueba
Pregunta: ¿Qué significa el # en vez del $?
3) Es una versión tan básica de Ubuntu que prácticamente nada viene pre-instalado, y si sí, no en su última versión. Por eso es buena idea correr apt-get update:
# apt-get update
Ign http://archive.ubuntu.com trusty InRelease
Get:1 http://archive.ubuntu.com trusty-updates InRelease [65.9 kB]
Get:2 http://archive.ubuntu.com trusty-security InRelease [65.9 kB]
Hit http://archive.ubuntu.com trusty Release.gpg
Hit http://archive.ubuntu.com trusty Release
Get:3 http://archive.ubuntu.com trusty-updates/main Sources [328 kB]
Get:4 http://archive.ubuntu.com trusty-updates/restricted Sources [5217 B]
Get:5 http://archive.ubuntu.com trusty-updates/universe Sources [190 kB]
Get:6 http://archive.ubuntu.com trusty-updates/main amd64 Packages [910 kB]
Get:7 http://archive.ubuntu.com trusty-updates/restricted amd64 Packages [23.5 kB]
Get:8 http://archive.ubuntu.com trusty-updates/universe amd64 Packages [440 kB]
Get:9 http://archive.ubuntu.com trusty-security/main Sources [134 kB]
Get:10 http://archive.ubuntu.com trusty-security/restricted Sources [3920 B]
Get:11 http://archive.ubuntu.com trusty-security/universe Sources [39.1 kB]
Get:12 http://archive.ubuntu.com trusty-security/main amd64 Packages [542 kB]
Get:13 http://archive.ubuntu.com trusty-security/restricted amd64 Packages [20.2 kB]
Get:14 http://archive.ubuntu.com trusty-security/universe amd64 Packages [162 kB]
Get:15 http://archive.ubuntu.com trusty/main Sources [1335 kB]
Get:16 http://archive.ubuntu.com trusty/restricted Sources [5335 B]
Get:17 http://archive.ubuntu.com trusty/universe Sources [7926 kB]
Get:18 http://archive.ubuntu.com trusty/main amd64 Packages [1743 kB]
Get:19 http://archive.ubuntu.com trusty/restricted amd64 Packages [16.0 kB]
Get:20 http://archive.ubuntu.com trusty/universe amd64 Packages [7589 kB]
Fetched 21.5 MB in 27s (778 kB/s)
Reading package lists... Done
5) Y ahora sí, podemos instalar herramientas, por ejemplo curl:
# apt-get install curl
5) Cuando termines puedes salir (exit) de este contenedor. Los cambios que hayas hecho no se guardarán en la imagen, pero sí en el contenedor que se creó al correrla.
Vamos a ver qué contenedores tenemos:
$ docker ps -a
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
8f36b66384cb ubuntu "bash" 5 minutes ago Exited (0) 3 minutes ago eager_brattain
3dd0d21a78b9 ubuntu "bash" 10 minutes ago Up 10 minutes gifted_hypatia
e3591a0c4077 ubuntu "bash" 14 minutes ago Exited (0) 13 minutes ago keen_clarke
1dab978ee569 hello-world "/hello" 16 minutes ago Exited (0) 16 minutes ago
Nuestro contenedor es el último. Para volver a entrar a él utilizamos primero start y su ID (el texto alfanumérico es el ID del container) y luego exec:
$ docker start 8f36b66384cb
8f36b66384cb
Ahora aparecerá en la lista de contenedores activos (docker ps SIN -a.
$ docker ps
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
8f36b66384cb ubuntu "bash" 8 minutes ago Up 42 seconds eager_brattain
Y por lo tanto podemos volver a entrar a el con exec
$ docker exec -it 8f36b66384cb bash
root@8f36b66384cb:/#
Nota que los cambios que hayas realizado dentro del contenedor continúan existiendo.
root@8f36b66384cb:/# ls
Prueba bin boot dev etc home lib lib64 media mnt opt proc root run sbin srv sys tmp usr var
Si nos salimos (exit) y luego queremos detenerlo por completo:
$ docker stop a5864268eadd
Si quieres que tu contenedor corra en el background desde un principio, puedes utilizar el flab -d.
$ docker run -dit ubuntu bash
e236ce647ea0c6513531f44f9880bc3b4c0005f2442dd16ca10125bd305ec31b
$ docker ps
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
e236ce647ea0 ubuntu "bash" 12 seconds ago Up 10 seconds hopeful_goodall
Borrar contenedores e imágenes
Si quieres borrar contenedores o imágenes (son espacio en disco):
-
Borrar un contenedor: Primero deterlo con
docker stop CONTAINER_IDy luego borrarlo condocker rm CONTAINER_ID -
Borrar una imagen:
docker rmi -f IMAGE_ID
“Conectar” un contenedor con un directorio dentro del OS nativo, aka “montar un volumen”:
Ejemplo con una imagen de Ubuntu.
1) Enlista las imagenes para asegurarte tener una de Ubuntu:
$ docker images
REPOSITORY TAG IMAGE ID CREATED SIZE
miproyecto/analisis1 v1 b951cd1b24b5 12 hours ago 188 MB
ubuntu latest 07c86167cdc4 13 days ago 188 MB
hello-world latest 690ed74de00f 5 months ago 960 B
Ya debes tener una imagen de ubuntu (si seguiste las notas anteriores), si no docker pull ubuntu.
3) Corre la imagen de ubuntu dentro de un contenedor, pero montando un volumen, es decir un directorio en tu equipo que podrá ser accedido por el contenedor:
docker run -v /home/cirio/aliciamstt/BioinfinvRepro/Unidad4/Prac_Uni4/DatosContenedor1:/DatosContenedorEjercicioClase -it ubuntu bash
Desglozando el comando anterior:
-v es la bandera para indicar que queremos que monte un volumen
/home/cirio/aliciamstt/BioinfinvRepro/Unidad4/Prac_Uni4/DatosContenedor1 es la ruta absoluta. Sí, absoluta (así que cambiala por la ruta de tu equipo o usuario) ya que así es cuando se trata de montar volúmenes :(.
:/DatosContenedorEjercicioClase es el nombre del directorio como quremos que aparezca dentro de nuestro contenedor. El :/ es para indicar que lo que sigue es el nombre.
4) Explora el volumen que montaste, prueba hacer un archivo. Nota que puedes acceder a el desde tu explorador, es decir todo lo que suceda en ese directorio puedes verlo/modificarlo desde dentro y fuera del contenedor.
root@dd4667e94adb:/# ls
DatosContenedorEjercicioClase bin boot dev etc home lib lib64 media mnt opt proc root run sbin srv sys tmp usr var
root@dd4667e94adb:/# cd DatosContenedorEjercicioClase/
root@dd4667e94adb:/DatosContenedorEjercicioClase# ls
eg_ddRAD_data.fastq
root@dd4667e94adb:/DatosContenedorEjercicioClase# touch Prueba
root@dd4667e94adb:/DatosContenedorEjercicioClase# ls
Prueba eg_ddRAD_data.fastq
Vamos a ver un ejemplo en el contenedor de Ubuntu donde montamos el volumen en el ejemplo de funcionamiento de Docker. Esto nos permitirá trabajar todos sobre lo mismo (y no romper nada):
Recuerda primero buscar el ID del contenedor (el ID será diferente en tu computadora) con docker ps -a. Luego:
$ docker start 8509d265036f
$ docker exec -it 8509d265036f /bin/bash
root@8509d265036f:/#
Empecemos por actualizar el software de ubuntu instalado de base y bajar algunos escenciales que no vienen en nuestra imagen de ubuntu down to basics.
apt-get update
apt-get install build-essential
Ejercicio:
Ve a la página de FastX-Tools y baja el archivo fastx_toolkit-0.0.14.tar.bz2 (ojo NO las librerías pre-compiladas). Guarda este archivo en DatosContenedor1 para que podamos verlo en el volumen DatosContenedorEjercicioClase.
7) Descomprime el archivo que acabas de bajar desde adentro de tu contenedor
tar -xvf fastx_toolkit-0.0.14.tar.bz2
(no pego el output de tar por brevedad) Pero aquí el resultado:
root@3c3c2e063371:/DatosContenedorEjercicioClase# ls
datos fastx_toolkit-0.0.14 fastx_toolkit-0.0.14.tar.bz2
root@3c3c2e063371:/DatosContenedorEjercicioClase# cd fastx_toolkit-0.0.14
root@3c3c2e063371:/DatosContenedorEjercicioClase/fastx_toolkit-0.0.14# ls
AUTHORS ChangeLog Makefile.am NEWS THANKS build_scripts config.h.in configure.ac galaxy m4 scripts
COPYING INSTALL Makefile.in README aclocal.m4 config configure doc install_galaxy_files.sh reconf src
8) Ejercicio Instala FastX-Tools dentro de un contenedor
5.5. Biocontainers
Dockerfiles
Recordemos que los contenedores de docker son creados a partir de una imagen. Dicha imagen puede ser básica, es decir el OS en su forma más simple, pero también puede incluir un software o conjunto de softwares ya instalados y listos para correr, y hasta los comandos que queremos que corra.
Lo anterior se hace a través de un dockerfile, es decir un script que describe (e instala) el software que pondremos en una imagen y además incluye cualquier detalle de la configuración del ambiente y hasta los comandos a correr.
Es decir un dockerfile nos permite construir y compartir una imagen especializada en correr el proceso que deseemos.
Veamos un dockerfile como este que instala el programa FastXTools que instalamos desde cero en el ejercico anteior.
Biocontainers
El dockerfile del ejercicio anterior vive en el github de Biocontainers. Y además en DockerHub de Biocontainers, por lo que podemos hacer un pull desde ahí.
Como verás, Biocontainers es una comunidad que se dedica a crear imágenes para instalar ese software que tanto necesitas. Checa sus contenedores.
La imagen base de Biocontainers se llama biocontainers y es un ubuntu más varias herramientas básicas de bioinformática. Checa su dockerfile aquí.
Vamos a bajar la imagen y correr un contenedor:
$ docker pull biocontainers/biocontainers
$ docker run -it biocontainers/biocontainers /bin/bash
biodocker@4c1831e1f7f7:/data$
$ curl
curl: try 'curl --help' or 'curl --manual' for more information
Observaciones y preguntas:
-
En vez de ser root (´#´ al inicio de la línea de comando) como es el default de docker, somos un usuario normal y estamos en un directorio llamado ´data´. ¿Con qué líneas del dockerfile se realizó esto?
-
curlestá instalado (el default no es este, según vimos la clase pasada). ¿Con qué líneas del dockerfile se especificó esto? -
Pude hacer un docker pull porque el dockerfile de arriba existe en un repositorio de contenedores llamado
biocontainers. -
La línea 100 del dockerfile dice
VOLUME ["/data", "/config"]¿Qué significa esto?
Ejercicio ¿Cómo puedo utilizar docker run para que el volumen data corresponda a un directorio en mi computadora?
Imagenes de sofware bioinformáico
Pero además del contendor básico biocontainers tenemos contenedores con ese sofware que siempre deseamos instalar. Por ejemplo con FastX-Tools y otro con vcftools. Vamos a hacer un pull:
$ docker pull biocontainers/fastxtools:0.0.14
$ docker pull biocontainers/vcftools:0.1.15
Yo puedo entrar a estos contenedores con -it /bin/bash como lo hemos hecho antes, pero también puedo utilizarlo para solo correr el programa con un comando concreto. Por ejemplo, mostrar la ayuda:
$ docker run --rm -u 1600 biocontainers/vcftools:0.1.15 vcftools -help
VCFtools (0.1.14)
© Adam Auton and Anthony Marcketta 2009
Process Variant Call Format files
For a list of options, please go to:
https://vcftools.github.io/man_latest.html
Alternatively, a man page is available, type:
man vcftools
Questions, comments, and suggestions should be emailed to:
vcftools-help@lists.sourceforge.net
o en FastX-tools:
$ docker run --rm -u 1600 biocontainers/fastxtools:0.0.14 fastq_to_fasta -h
usage: fastq_to_fasta [-h] [-r] [-n] [-v] [-z] [-i INFILE] [-o OUTFILE]
Part of FASTX Toolkit 0.0.14 by A. Gordon (assafgordon@gmail.com)
[-h] = This helpful help screen.
[-r] = Rename sequence identifiers to numbers.
[-n] = keep sequences with unknown (N) nucleotides.
Default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA output file. default is STDOUT.
- Nota: la flag
-u 1600sirve para correr el contenedor como usuarios raiz en vez del usuario de biodocker. Esto es necesario para correrlo en el cluster de la conabio por como tiene la configuración de los permisos, pero no en tu computadora.
Ejercicio: ve a la página https://biocontainers-edu.biocontainers.pro/en/latest/running_example.html y lee el ejemplo de cómo usar blast. Escribe un script para adoptar el ejemplo de esta página a tu computadora. Guarda tu script en tu repositorio de Github para las tareas del curso y brinda el link a dicho script.
Tip: borrar automáticamente un contenedor cuando acaba de correr
Cada vez que utilizamos docker run se crea un contendor nuevo. Por ejemplo si corriste los docker run de fastxtools y vcftools de arriba sin la flag --r:
$ docker ps -a
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
5750fafe938a biocontainers/vcftools "vcftools -help" 2 seconds ago Exited (0) 3 seconds ago practical_yonath
4845c81d1dc6 biocontainers/fastxtools "fastq_to_fasta -h" 17 seconds ago Exited (1) 17 seconds ago elastic_villani
Esto ocupa espacio en disco y nos llena de contenedores a los que no volveremos a entrar. La solución: borrar un contenedor al salir, utilizando el flag --rm
Si quieres correr varios comandos juntos en un mismo contenedor, necesitas agregar --rm. Más una de estas dos opciones:
1) Indicandole al contenedor que se salga al terminar de correr, agregando -c al final de los comandos que queremos que corra. Ejemplo:
$ docker run -u 1600 --rm biocontainers/fastxtools:0.0.14 fastq_to_fasta -h -c
usage: fastq_to_fasta [-h] [-r] [-n] [-v] [-z] [-i INFILE] [-o OUTFILE]
Part of FASTX Toolkit 0.0.14 by A. Gordon (assafgordon@gmail.com)
[-h] = This helpful help screen.
[-r] = Rename sequence identifiers to numbers.
[-n] = keep sequences with unknown (N) nucleotides.
Default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA output file. default is STDOUT.
$ docker ps -a
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
2) El flag -c en realidad sirve para pedirle que corra más de un comando dentro del mismo contenedor (unidos por ejemplo con |, ;, etc). Si corres el contenedor con bash -c y los comandos deseados entre ““ automáticamente se saldrá. Ejemplo:
$ docker run -u 1600 --rm biocontainers/fastxtools:0.0.14 bash -c "fastq_to_fasta -h ; echo hola mundo"
usage: fastq_to_fasta [-h] [-r] [-n] [-v] [-z] [-i INFILE] [-o OUTFILE]
Part of FASTX Toolkit 0.0.14 by A. Gordon (assafgordon@gmail.com)
[-h] = This helpful help screen.
[-r] = Rename sequence identifiers to numbers.
[-n] = keep sequences with unknown (N) nucleotides.
Default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA output file. default is STDOUT.
hola mundo
$ docker ps -a
CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES
Si te quedan dudas sobre Docker y cómo aplicarlo a Bionformática revisa esta excelente sección de ayuda de Biocontainers.
Repaso y conceptos clave
- Utilidad de docker:
1 ) Máxima reproducibilidad, al igualar OS y versiones de un software.
2) No romper tu equipo/otroprograma tratando de instalar algo
-
Docker permite correr un programa en un contenedor a partir de una imagen que podemos
pulldesde un repositorio online, como Biocontainers o DockerHub. -
Dentro de un contenedor está el OS, las librerías y otros requisitos que requiera un programa y el programa mismo.
-
Para “conectar” un contenedor de Docker con el mundo necesitas montar un volumen:
docker run -v [RutaABSOLUTAaldirectorioDeseado:/nombrevolumen] [imagen] [comandos a correr]. -
Formas para correr un software bioinformático usando Docker:
1) Entrando a un contenedor con todo lo que queremos: suponiendo que en un contenedor vayas instalando todo el sofware que necesitas para un análisis dado. Los comandos base que necesitas para esto serían:
docker pull [IMAGEN] para bajar la imagen base donde trabajaras
docker run -v [-v [RutaABSOLUTAaldirectorioDeseado:/nombrevolumen]] -it [IMAGEN] bash para crear y correr el contenedor con la imagen de forma interactiva (“entrando”) y con un volumen montado a un directorio de nuestra compu donde queramos escribir/leer datos. Ejemplo: docker run -v /home/cirio/aliciamstt/BioinfinvRepro/Unidad4/Prac_Uni4/DatosContenedor1:/DatosContenedorEjercicioClase -it ubuntu bash
OJO: cada vez que haces docker run se crea un contenedor distinto a partir de la misma imagen. Para no llenarte de contenedores utiliza docker run --rm (detalles abajo).
docker restart IDCONTAINER para prender de nuevo el contenedor, NO docker pull de nuevo.
docker exec -it IDCONTAINER bash para volver a entrar al contenedor.
docker rm para borrar un contenedor que ya no quieras. Debes docker stop si está corriendo.
2) Utilizando un contenedor por proceso (Recomendado) suponiendo que el sofware que utilizas ya vive en una imagen, por ejemplo de Biocontainers. Los comandos base que utilizarías para esto serían:
docker pull [biocontainers/IMAGEN] para bajar la imagen de cada sofware que utilizarás, por ejemplo de Biocontainers.
docker run --rm -v [RutaABSOLUTAaldirectorioDeseado:/data] [biocontainers/IMAGEN] [COMANDOS del sofware en cuestión] -c para correr el contenedor de una imagen de biocontainers con los comandos específicos de un software dado, con volumen montado a un directorio de nuestra compu donde queramos escribir/leer datos y de tal forma que el contenedor se borre automáticamente al terminar el proceso. Ejemplo:
docker run -u 1600 --rm -v /home/cirio/aliciamstt/BioinfinvRepro/Unidad4/Prac_Uni4/DatosContenedor1/datos:/data biocontainers/fastxtools:0.0.14 bash -c "fastx_trimmer -f 1 -l 70 -i human_Illumina_dataset.fastq -v | fastq_quality_filter -q 20 -p 90 -o clean_human_data.fastq -v"
(Estos datos ejemplo vienen de Galaxy Data Libraries y son de libre uso)
RECOMENDACIONES
-
En lugar de poner líneas tan largas como las de arriba párte comando por comando utilizando
\, que es como;pero puedes dar enter y continuar en la línea de abajo. -
Declara una variable con la ruta absoluta al inicio de tu script y luego utilizala dentro de tu línea de
docker run -v.
Ejercicio: corre el ejemplo de limpiar human_Illumina_dataset.fastq utilizando una variable llamada miruta con la ruta absoluta a DatosContenedor1/datos dentro de tu directorio del cluster o en tu equipo
Código a modificar:
docker run -u 1600 --rm -v /home/cirio/aliciamstt/BioinfinvRepro/Unidad4/Prac_Uni4/DatosContenedor1/datos:/data biocontainers/fastxtools:0.0.14 bash -c "fastx_trimmer -f 1 -l 70 -i human_Illumina_dataset.fastq -v | fastq_quality_filter -q 20 -p 90 -o clean_human_data.fastq -v"
Recuerda que el flag -u 1600 sólo es necesario si lo corres en el cluster de la CONABIO.