BioinfinvRepro
Curso de introducción a la bioinformática e investigación reproducible
Tutorial para el filtro y alineamiento de lecturas
Karen Oróstica y Cristian Yañez Ingenieros en Bioinformática Abril 2020
En este tutorial presentamos los pasos necesarios para realizar el filtrado y alineamiento de secuencias. En la siguiente imagen les mostramos el pipeline a seguir en este tutorial. El análisis de calidad fue realizado en el la clase anterior.
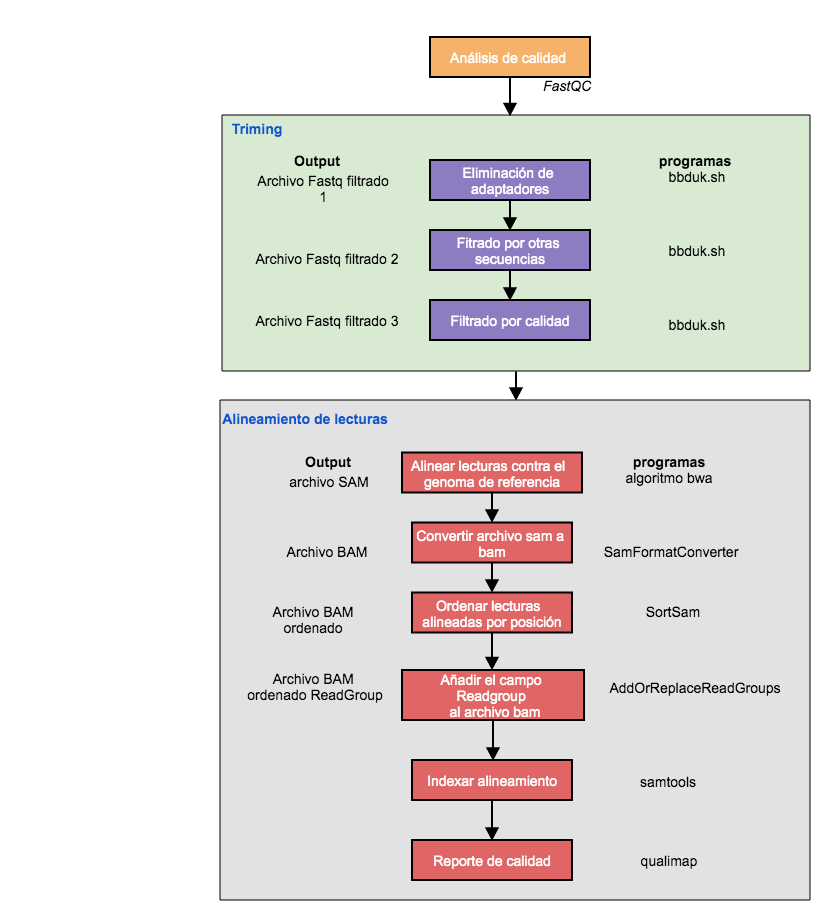
El siguiente protocolo está diseñado para ser implementado en el servidor genoma.med.uchile.cl dentro de la carpeta de cada usuario de bioinfo1.
Conectarse al servidor
ssh -Y bioinfo1@genoma.med.uchile.cl
Usar la clave entregada en clase.
Acceda al directorio con su nombre:
cd jperez/
Para la demostración, usaremos el directorio korostica. El alumno debe usar su propio directorio.
1. Filtrado de lecturas
Las lecturas crudas pueden contener secuencias de adaptadores usados en la secuenciación, contaminantes y sitios con bajas calidades. Comúnmente en las técnicas NGS el extremo 3’ posee una menor calidad que el extremo 5’. El procesamiento de las lecturas para eliminar las secuencias pertenecientes a adaptadores y los pares de bases con baja calidad se denomina Trimming. Para realizar un trimming adecuado de lecturas se debe hacer un análisis previo de la calidad de la secuenciación mediante el programa FastQC. Una vez realizado este análisis se realiza un trimming de lecturas usando el programa bbduk en 3 pasos:
1.1 Eliminación de adaptadores
Comando:
bbduk.sh -Xmx2g threads=1 \
in1=muestra_R1_001.fastq.gz \
in2=muestra_R2_001.fastq.gz \
out1=muestra_R1_filter1.fastq.gz \
out2=muestra_R2_filter1.fastq.gz \
ref=adapters.fa tpe tbo
Ejemplo:
/opt/bbmap/bbduk.sh -Xmx2g threads=1 \
in1=../181004_curso_calidad_datos_NGS/fastq_raw/S10_R1.fastq.gz \
in2=../181004_curso_calidad_datos_NGS/fastq_raw/S10_R2.fastq.gz \
out1=S10_R1_filter1.fastq.gz \
out2=S10_R2_filter1.fastq.gz \
ref=/opt/bbmap/resources/adapters.fa tpe tbo
La entrada in1, in2 son las lecturas crudas. Esto crea dos archivos con el sufijo “filter1.fastq.gz” correspondiente a las lecturas pareadas donde se eliminan la secuencia de adaptadores y contaminantes que alinean con las secuencias del archivo fasta “adapters.fa”.
1.2 Eliminación de lecturas que contengan secuencia del fago phix
Comando:
bbduk.sh -Xmx2g threads=1 \
in1=muestra_R1_filter1.fastq.gz \
in2=muestra_R2_filter1.fastq.gz \
out1=muestra_R1_filter2.fastq.gz \
out2=muestra_R2_filter2.fastq.gz \
ref=phix174_ill.ref.fa.gz
Ejemplo:
/opt/bbmap/bbduk.sh -Xmx2g threads=1 \
in1=S10_R1_filter1.fastq.gz \
in2=S10_R2_filter1.fastq.gz \
out1=S10_R1_filter2.fastq.gz \
out2=S10_R2_filter2.fastq.gz \
ref=/opt/bbmap/resources/phix174_ill.ref.fa.gz
La entrada (in1, in2) son las lecturas filtradas en el paso anterior. Esto crea un 2 archivos “filter2.fastq.gz” correspondiente a las lecturas pareadas donde se eliminan la secuencia del fago phix que alinean con las secuencias del archivo fasta “phix174_ill.ref.fa.gz”.
1.3 Filtrado por calidad de las lecturas
Comando:
bbduk.sh -Xmx2g threads=1 \
in1=muestra_R1_filter2.fastq.gz \
in2=muestra_R2_filter2.fastq.gz \
out1=muestra_R1_filter3.fastq.gz \
out2=muestra_R1_filter3.fastq.gz \
qtrim=w trimq=20 minlength=30 minavgquality=20
Ejemplo:
/opt/bbmap/bbduk.sh -Xmx2g threads=1 \
in1=S10_R1_filter2.fastq.gz \
in2=S10_R2_filter2.fastq.gz \
out1=S10_R1_filter3.fastq.gz \
out2=S10_R2_filter3.fastq.gz \
qtrim=w trimq=20 minlength=30 minavgquality=20
La entrada (in1, in2) son las lecturas filtradas en el paso anterior. Estas lecturas son filtradas por calidad usando una ventana deslizante con el parámetro “qtrim=w”, que elimina la región de la lectura con una calidad menor a 20 en adelante “trimq=20” (hacia el extremo 3’), luego evalúa el largo resultante de la lectura, eliminandola si es menor a 30 pb “minlength=30”. Como último filtro, elimina la lectura si el promedio de calidad total es <= 20 “minavgquality=20”. De esta forma se obtienen las lecturas filtradas (“filter3”). Estos parámetros fueron establecidos como óptimos al analizar las lecturas crudas con el programa FastQC, pero pueden ser modificados acorde a la calidad de los datos.
2. Alineamiento de lecturas
2.1 Alinear lecturas contra el genoma de referencia
Se alinean las lecturas contra el genoma de referencia, obteniéndose un archivo SAM (Sequence alignment map).
Comando:
bwa mem -t 4 -M reference.fasta \
muestra_R1.fastq.gz \
muestra_R2.fastq.gz \
> muestra.sam
Ejemplo:
bwa mem -t 4 -M /home-old/data/references/genomes/hg19_reference/hg19.fasta \
S10_R1_filter3.fastq.gz \
S10_R2_filter3.fastq.gz \
> S10.sam
Nota 1: Revise la documentación de bwa e inspeccione las distintas opciones que entrega bwa y los atributos que puede modificar.
El siguiente conjunto de operaciones de preprocesamiento formatea los datos para adaptarse a los requisitos de las herramientas GATK convirtiendo los datos de asignación en un archivo BAM ordenado por posición, con el campo “Read Group” añadido.
2.2 Convertir archivo sam a bam (binario)
Se obtiene un bam (binary alignment map) a partir del alineamiento en formato sam.
Comando:
java -jar picard.jar SamFormatConverter \
I=muestra.sam \
O=muestra.bam
Ejemplo:
java -jar /opt/picard-tools-2.5.0/picard.jar SamFormatConverter I=S10.sam O=S10.bam
2.3 Ordenar lecturas alineadas por posición
Obtenemos un archivo bam con las lecturas ordenadas por posición del alineamiento a la referencia.
Comando:
java -jar picard.jar SortSam \
I=muestra.bam \
O=muestra_sorted.bam \
SO=coordinate
Ejemplo:
java -jar /opt/picard-tools-2.5.0/picard.jar SortSam I=S10.bam O=S10_sorted.bam SO=coordinate
2.4 Añadir el campo Readgroup al archivo bam
Comando:
java -jar picard.jar AddOrReplaceReadGroups \
I=muestra_sorted.bam \
O=muestra_sorted_RG.bam \
ID=muestra LB=Paired-end PL=Illumina PU=Unknown SM=muestra
Ejemplo:
java -jar /opt/picard-tools-2.5.0/picard.jar AddOrReplaceReadGroups I=S10_sorted.bam O=S10_sorted_RG.bam ID=sample LB=Paired-end PL=Illumina PU=Unknown SM=sample
2.5 Indexar alineamiento
Comando:
samtools index muestra_sorted_RG.bam
Ejemplo:
samtools index S10_sorted_RG.bam
2.6 Generar un reporte de calidad
Qualimap es una aplicación escrita en Java y R que posee una interfaz gráfica de usuario (GUI) y una interfaz de línea de comandos para facilitar el control de calidad de los datos de alineamiento de secuencias. Este programa proporciona una vista general de los datos que ayuda a detectar sesgos en la secuenciación y / o mapeo de los datos y facilita la toma de decisiones para un análisis posterior.
Comando:
qualimap bamqc -bam muestra_sorted_RG.bam -gff regiones_blanco.bed
Ejemplo:
/opt/qualimap_v2.2.1_2/qualimap bamqc -bam S10_sorted_RG.bam -gff ../181004_curso_calidad_datos_NGS/regiones_blanco.bed -outdir ./S10_sorted_RG
3 Tarea
1) Realizar el alineamiento de las lecturas R1 y R2 del paciente seleccionado para la tarea anterior contra el genoma humano hg19. 2) Generar un reporte técnico de calidad del alineamiento con qualimap. 3) Seleccionar 4 figuras que a su juicio sean las más informativas sobre la calidad de los datos y del ensamble. 4) Incluir las figuras en la sección de Resultados de un reporte técnico. Describir cada figura con una leyenda descriptiva. Adicionalmente, en el texto de la sección, interpretar los resultados y citar cada figura. Debe referirse a la calidad de los datos y del alineamiento. Enfóquese especialmente en los posibles problemas con los datos o alineamientos. Comente potenciales razones que expliquen lo observado. Incluya una sección con las principales Conclusiones para la muestra. 5) Incluya el reporte completo generado con qualimap como anexo. 6) Envíe el informe mediante Google classroom, al dar por completada esta tarea.
Anexo
Los pasos descritos en este anexo son necesarios previo a la ejecución de los procedimiento descritos en el tutorial y ya fueron realizados para facilitar su desarrollo en nuestro servidor. Si usted realiza estos análisis en otro servidor, necesitará realizar estos pasos previamente.
1. Preparar la referencia
El genoma humano de la referencia usado para el procesamiento de las lecturas es hg19. Para usar este genoma en formato fasta se debe procesar para generar distintos archivos necesarios para el flujo de trabajo.
Procesamiento 1 (necesario para usar el programa BWA):
Comando:
bwa index -a bwtsw reference.fasta
Procesamiento 2 (necesario para usar el programa BWA):
Comando:
samtools faidx reference.fasta
Procesamiento 3 (necesario para usar el programa GATK):
Comando:
java -jar picard.jar CreateSequenceDictionary REFERENCE=reference.fasta OUTPUT=reference.dict
Nota 1: La preparación de la referencia se debe realizar solo una vez, para efectos prácticos del curso la referencia ya se encuentra procesada con los comandos descritos.